

Dr. Paul G. Harch discusses hyperbaric oxygen therapy (HBOT) as a generic drug for reperfusion injury, after presentation to the 5th European Federation of Neurological Societies Meeting in Copenhagen, Denmark, 2000
An entry from K.K. Jain’s Textbook Of Hyperbaric Medicine
Hyperbaric oxygen therapy has been used in a number of conditions characterized by global ischemia (as opposed to focal ischemia of stroke), and anoxia, and leading to impairment of consciousness. Conditions such as coma due to brain injury and anoxia associated with drowning and hanging are discussed under the following headings:
Pathophysiology
Rational Basis of HBOT Therapy
Review of Animal Experimental Studies
Review of Human Clinical Studies
Case Studies
Conclusions
Introduction
For a discussion of the effectiveness of hyperbaric oxygen (HBOT) therapy in global cerebral ischemia/anoxia and coma, we define HBOT as a medical treatment that uses high pressure oxygen as a drug by fully enclosing a person or animal in a pressure vessel and then adjusting the dose of the drug to treat pathophysiologic processes of the diseases. Like all drugs, the dose of HBOT is crucial and should be customized to each patient’s response. It is dictated by the pathological target and is determined by the pressure of oxygen, duration of exposure, frequency, total number of treatments, and timing of the dose in the course of the disease. As diseases and their pathologies evolve, different doses of HBOT are required at different times. In addition, patients have individual susceptibility to drugs, manifest side effects and toxicity. Unfortunately, the ideal dose of HBOT in acute or chronic global ischemia/anoxia and coma is unknown. The studies reviewed below suggest higher pressures (2 ATA or higher) and lesser numbers of treatments very early in the disease process whereas lower pressures (2 ATA or lower) and a greater number of treatments have been used as the brain injury matures. While this general trend seems justified, the absolute or effective pressures delivered to the patients in these reports may be slightly less than what is stated since many studies do not specify the HBOT delivery system that was employed. For example, an oxygen pressurized chamber has an effective HBOT pressure equal to the plateau pressure administered during the treatment, whereas an air pressurized chamber in which oxygen is administered by aviators mask can achieve a far lower effective HBOT pressure, depending on the fit of the mask and the amount of its air/oxygen leak. In the later cases, the dose of oxygen is less. This concept is particularly important when analyzing the studies in this chapter performed prior to the late 1980s when the aviator mask dominated delivery systems in multiplace chambers.
In reviewing the data in this chapter, it is surprising that HBOT has not enjoyed widespread use for neurological diseases in the United States. This has been partly due to institutional reservations and overt therapeutic nihilism for neurological injuries, both of which are presently waning. To assume that HBOT could have efficacy and benefit when liberally applied to various “accepted” indications, yet have none in the great majority of neurological conditions is perplexing. After all, the brain is enclosed within the same body in the same pressure vessel and is exposed to the same elevated oxygen pressure. To justify this distinction, one would have to postulate that an entire set of pathophysiological processes of brain that are insensitive to HBOT and distinct from those in the rest of the body’s organ systems which are sensitive to HBOT and to which we routinely apply HBOT. This is illogical and unlikely. Such reasoning is indefensible when one considers the “accepted” neurological indications include carbon monoxide poisoning, brain decompression sickness, cerebral air embolism, brain abscess, and cyanide poisoning. We conclude that HBOT should benefit other hypoxic/ischemic conditions of the brain, provided the dose is correct, i.e., target specific.
Other reasons for non-recognition of HBOT in neurological conditions concern methodologies. The standard for proof in scientific medicine has been the randomized prospective controlled double-blinded clinical trial. While some of the studies in this chapter meet this rigor (except for double-blinding), many do not. Some are randomized, prospective, and controlled and thus exceed the quality of studies used to sanction reimbursement for some HBOT indications. Other studies are uncontrolled series, case-controlled, or individual cases. All of this clinical data, in conjunction with the animal data, makes a strong case for at least attempting HBOT in what are otherwise untreatable conditions with debilitating, tragic, and expensive outcomes, especially when the visual medium is used to prove single-case causality (Kiene & von Schon-Angerer 1998; Harch 1996). In addition, case-controlled series with chronic neurological maladies make powerful statements of efficacy from the statistical (Glantz 1992) and logical perspectives where the counterargument of placebo effect is minimized (Kienle & Kiene 1996). If these considerations are kept in mind when analyzing this chapter, it appears that the bulk of data is solidly in favor of a beneficial effect of HBOT in global ischemia/anoxia and coma.
Pathophysiology
The effect of global ischemia/hypoxia on the brain has been discussed in Chapter 5. Oxidation of glucose is the primary energy source for the brain. Deprivation of oxygen causes deep psychological unresponsiveness in 8 s while glucose and energy stores take a few minutes to exhaust (Plum & Pulsinelli 1992). Global deprivation of oxygen delivery can be achieved by reduction in blood flow (ischemia), oxygen (hypoxia/anoxia), or both (hypoxic or anoxic ischemia). Unfortunately, clinical syndromes and animal models are rarely pure and often result from combinations or sequences and varying degrees and durations of hypoxia/anoxia and ischemia. Since the insult, oxygen deprivation, is similar whether by lowered blood flow or oxygen content the two are often considered as a single type of insult and this concept will be followed in this chapter.
Global ischemia/anoxia is a severe transient insult to the brain that causes a stereotypic pathophysiology characterized by reperfusion hyperemia followed by progressive ischemia which is often heterogeneous (Safar 1986; Dirnagl 1993). The extent of injury is governed by a complex interplay of patterns of systemic and local respiratory and circulatory function and selective vulnerability of cells (Meyers 1979). The result is both immediate and delayed cell death ( Cormio et al1997). The deterioration of blood flow and late cell death occur in the absence of microvascular disruption by formed blood elements unless the global ischemia is prolonged (Dirnagl1993) in which case both cellular and non-cellular mechanisms may be responsible. The mechanisms of eventual neuronal energy failure are poorly understood (Siesjo et al1995, Katsura et al1995).
Coma, on the other hand, is a neurological state resulting from a wide variety of cerebral insults that is caused by diffuse disruption (functional or anatomical) of the bilateral cerebral cortices, proximal brainstem (reticular activating system), or both (Rossor 1993). Coma is characterized by an alteration in the level of awakeness, ranges from mild somnolence to deep coma, and is graded on a number of scales, the best known of which is the Glasgow Coma Scale (Teasdale & Jennett 1974). In the studies reviewed below, coma usually refers to the more severe end of the continuum: unresponsiveness, posturing, and neurovegetative signs, however, a number of studies are unclear about the exact level of coma.
Rational Basis of HBOT Therapy
HBOT in acute global ischemia/anoxia is complicated by a lack of knowledge of the exact pathological targets and their oxygen sensitivity. It has been postulated that post-ischemic hypoperfusion may be a neurogenic reflex (Dirnagl 1993) and/or characterized by a block in the transduction of physiologic stimuli and hence protein synthesis (Siesjo 1981). Assuming short-lived (minutes) global ischemia/anoxia and cell death independent of the microcirculation (Dirnagl1993 ), positive effects of HBOT under these conditions must be due to effects on hypoxia, cellular energy metabolism, ion homeostasis, membrane integrity, gene induction, and/ or a plethora of as yet unidentified targets. The dramatic effect of even one HBOT exposure on recovery of brain function, as indicated in many of the studies below, implies a powerful on/off drug effect that simultaneously quenches a degradative process and energizes the cell. It is easy to envision HBOT acting at some or multiple points of blockade in the above mentioned reflex or at a physiologic impasse. In the chronic state, a similar action of a single HBOT on some of these targets may be responsible for the awakening of idling neurons (Neubauer et al 1990), and when delivered repetitively considered a signal transducer (Siddiqui 1995). The signal transduction mechanism is inferred in multiple non-cerebral HBOT wound models where trophic tissue changes result from repetitive HBOT (HBOT Committee Report 2003), measured molecularly in two studies (Wu etal1995, Buras etal2000), suggested in a replicated chronic traumatic brain injury rat model (Harch & Mustoe 1996), and reaffirmed in a controlled human trial of HBOT in chronic traumatic brain injury directed by Harch in Texas (Barrett et al1998). In the rat model repetitive 1.5 ATA/90 min HBOTT induced increased blood vessel density and cognitive function in injured hippocampus. This finding likely underpins much of the trophic effect of HBOTT in chronic brain injury and represents the first ever improvement of chronic brain injury in animals. Future studies should be focused on elucidating the molecular effects of HBOT in this model and in global cerebral ischemia/anoxia.
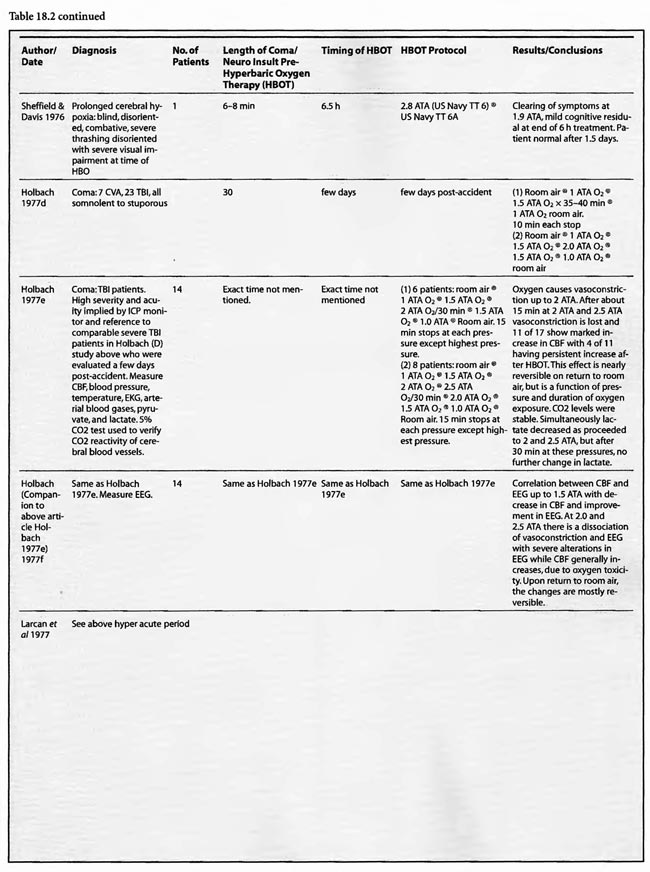
With prolongation of global ischemia/anoxia or the recovery phase of reperfusion pre-therapeutic intervention the microcirculation is disturbed (Dirnagl 1993) and the pathophysiology begins to resemble that in acute traumatic brain injury (Cormio et al1997): lipid peroxidation, edema, arterial spasm, cellular reperfusion injury, and anaerobic metabolism in the setting of penumbral lesions (Cormio et al1997). HBOT has been shown to have positive effects on all of these: ischemic penumbra (Neubauer et al 1990; Neubauer et al1998; Barrett et al1998), cerebral edema (Sukoff et al 1982), arterial spasm (Kohshi et al1993, Kawamura 1988), anaerobic metabolism (Holbach 1977d), and peroxidation/ cellular reperfusion injury (Thorn 1993a). This last HBOT sensitive pathophysiological target is most exciting since it seems to be a generic ischemic model-, species-, and organ-independent HBOT effect (Harch 2000). In a carbon monoxide rat model Thorn (1993b) showed a powerful inhibitory action of HBOT on white blood cell mediated brain lipid peroxidation when delivered 24 hours before the poisoning or 45 min after removal from carbon monoxide. Zamboni et al ( 1993) demonstrated a similar finding in a four hour global ischemic rat gracilis muscle model, using intravital microscopy. Follow-up work by Khiabani et al (2001) identified a plasma mediator(s) of this phenomenon. This HBOT inhibition of WBCs is inferred in brain decompression sickness and cerebral air embolism (Harch 1996) when one combines the Dutka et al (1989), Helps & Gorman (1991) and Martin (2001) data, which implicates WBCs in the pathogenesis of these disorders, with Thalmann’s (1990) review which shows a 90o/o single treatment cure rate in decompression sickness when hyperbaric recompression is delivered within the first 1-2 h of injury. Similarly, the data of Bulkley & Hutchins ( 1977) and Engler et al ( 1986) that document a WBC-mediated pathogenesis in cardiac reperfusion injury, in conjunction with the Thomas etal (1990) tissue plasminogen activator/HBOT/acute myocardial infarction dog model and the congruent human study of Shandling et al ( 1997) strongly suggest an HBOT directed action on cellular reperfusion injury, among other effects. Lastly, the recent Rosenthal paper (2003) demonstrates a positive effect on the microcirculation similar to the findings of Zamboni et al (1993). All of the above actions of HBOT on the pathophysiology in acute traumatic brain injury should sum to provide a beneficial effect. In fact, such is the case as a review of the studies in Table 18.2 shows a convincing argument for the use of HBOT in acute severe traumatic brain injury. Similarly, if global ischemia/anoxia is prolonged or incomplete, e.g., unsuccessful hanging, microcirculatory disturbances are incomplete. Under these circumstances, HBOT-induced inhibition of cellular reperfusion injury may partly explain the very positive results of the studies listed in Tables 18.1 and 18.2.
For HBOT to be effective in coma it must be directed at diffuse targets in the bilateral hemispheric gray and white matter, the brainstem, or both. Acutely, regardless of the nature of the targets, e.g., microcirculatory, non-vascular, cellular, or other, HBOT can conceivably act equally effectively on the hemispheres, brainstem, or both. As the pathology progresses to anatomic damage and eventually to penumbral lesions, a significant HBOT effect on hemispheric coma is very unlikely because of the large tissue volumes and low ratios of umbra to penumbra. Smaller tissue volumes are favored such that brainstem coma would be expected to have better results. This is suggested by the positive HBOT data in the large traumatic mid-brain report of Holbach (1974b), the brainstem contusion subgroup of Artru (1976), and the coma patients of Heyman etal (1966) and Neubauer (1985a). In all of these clinical trials, a recovery of just a few millimeters of reticular activating system can translate into far-reaching effects in the hemispheres, e.g., awakening. Additional work will be necessary to confirm this hypothesis.
In chronic global ischemia/anoxia and coma the pathological targets become more speculative. The ischemic penumbra (Astrup et al1981) argument of Neubauer and Gottlieb (1990) and sympathetic hibernating myocardium concept of Swift et al (1992) remain, but given the numbers of treatments reported below the element of time enters the equation and implies a trophic effect. This effect, which could include stimulation of axon sprouting, or possibly an alteration or redirection of blood flow as suggested above (Harch 1996), or both, may be indiscriminately effective on the final common pathway of a variety of brain injuries similar to HBOT’s generic effect on reperfusion injury (Harch 2000). Given the diversity of reports (Neubauer etal1998; Harch 1994a,b,c; Harch 1996; Keirn et al1997; Myers 1995a) and the multitude of studies listed below this maybe so. Regardless of the nature of this generic effect, in the past 17 years (PGH) and 33 years (RAN) these authors have noted a upward sawtooth response (Chapter 17 Stroke, Figures 17.7 and 17.9) during HBOT in sub-acute and chronic brain injury with a partial regression once each round of HBOT ends. This implies permanent and transient components to the treatment (see Chapter 41 Neuroimaging). While the net HBOT result appears uniform, the biochemical, molecular, cellular, and anatomic complexities of the transient, permanent, and final effects will need to be developed in the future.
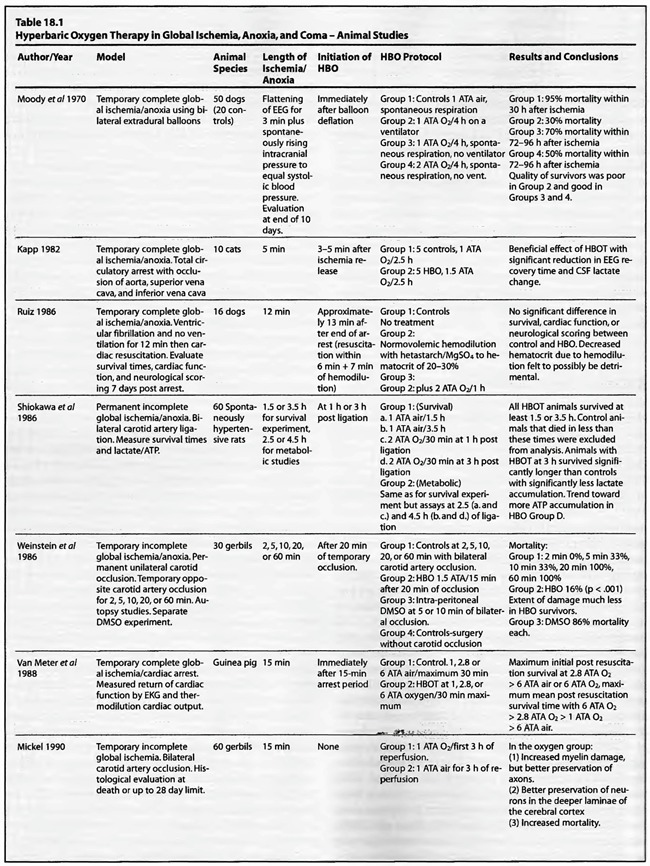
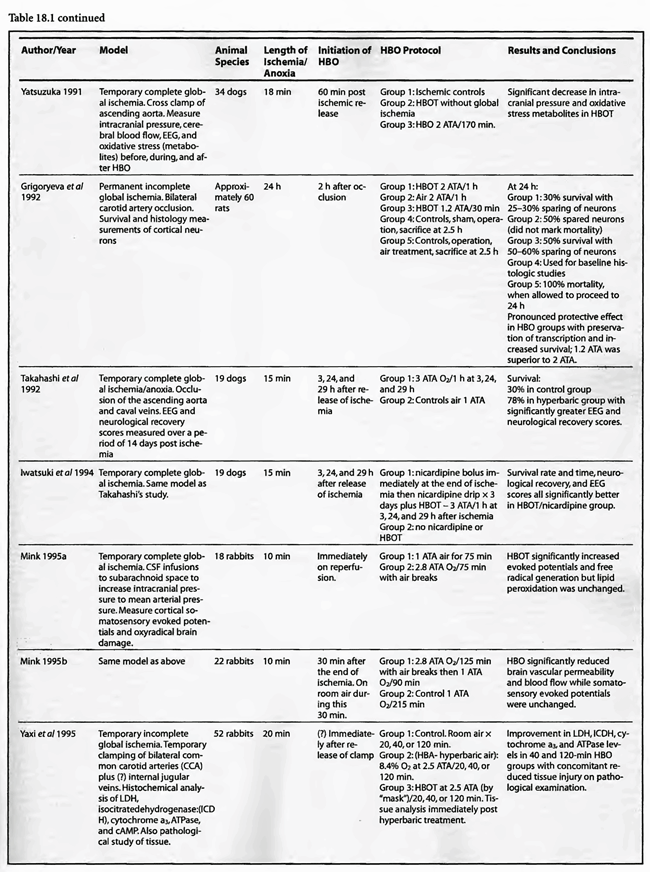
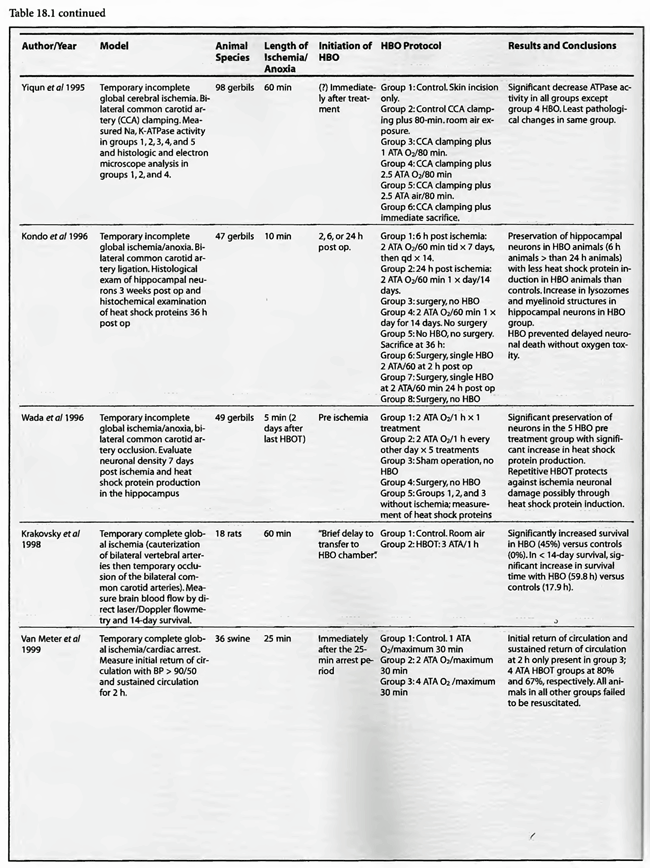
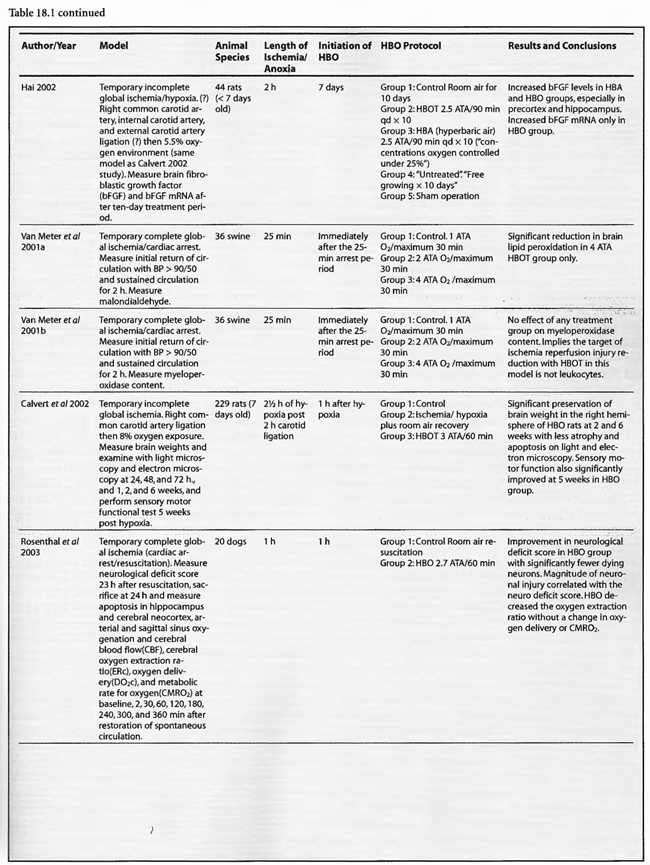
Review of Animal Experimental Studies
A review of the studies listed in Table 18.1leads to the conclusion that HBOT is unequivocally beneficial in acute global ischemia/anoxia regardless of treating pressure, frequency, duration, number of treatments, or time to onset of HBOT post insult. Since the first publication of this chapter in the third edition of the Textbook in 1999, the data has been fortified. Eight additional studies were added to the previous fourteen. Twenty of the twenty-two studies gave positive results, one study did not show any benefit, and the last study used normobaricoxygen. In the completed ischemia models, Moody et al (1970) showed a nearly 50% reduction in mortality without the benefit of artificial ventilation using a prolonged 2 ATA exposure (four hours). Kapp (1982) measured EEG recovery time and CSF lactate change, demonstrating significant improvement with a lower pressure, 1.5 ATA, for a prolonged 2.5 hours. Ruiz et al (1986) was the sole insignificant result with a 2 ATA/1 hour exposure, but this lack of efficacy may be partially explained by hemodilution with hetastarch prior to HBOT. Yatsuzuka (1991) generated a significant decrease in ICP and oxidative stress metabolites with a 2 ATA/ 170 min staged protocol. Using higher pressures, Takahashi et al (1992), Iwatsuki et al (1994), and Krakovsky et al (1998) at 3 ATA, Mink and Dutka (1995a,b) at 2.8 ATA, and Rosenthal et al (2003) at 2.7 ATA obtained statistically significant positive results on survival, neurological recovery, and various physiological or metabolic measures. In the Rosenthal experiment survival improved with increased oxygen extraction ratio but without a change in oxygen delivery or cerebral metabolic rate for oxygen, suggesting an improvement of the microcirculation similar to the Zamboni et al (1993) HBO/peripheral global ischemia reperfusion model in rats. The study by Mink and Dutka (1995b) had conflicting results with a simultaneous decrease in brain vascular permeability and blood flow while somatosensoryevoked potentials were unchanged. This implies concomitant beneficial and detrimental effects which are difficult to explain without more data. The four Van Meter et al studies (1988, 1999, 2001a, and 2001b) are unique in that they showed resuscitation of animals using HBOT, rather than delivering HBOT after resuscitation as in the Rosenthal article. These combined experiments were dose-response evaluations of HBOT at 1.0, 2.0, 2.8, 4.0, and 6.0 ATA. The swine study proved the ability of 4 ATA HBOT to resuscitate animals after 25 min of cardiac arrest, simultaneously truncating white blood cell-independent brain lipid peroxidation. This is the longest successful arrest/resuscitation reported in the medical literature and has profound implications for application to human cardiac arrest (see Chapter 37, HBOT in Emergency Medicine). In 10 of the 12 studies the benefit of HBOT was generated with one treatment, and in the other 2 studies with 3 treatments. No consensus emerges for the ideal dose ofHBO after complete global ischemia, but the Van Meter dose-response study suggests that HBOT at 4 to 6 ATA was effective in resuscitating animals with cardiac arrest. More importantly, in the twelve studies, a beneficial effect on global ischemia, anoxia, and coma was demonstrated even when the ischemic insult was as long as one hour (two studies) and the delay to treatment as long as six hours after the ischemic insult.
The results are similarly impressive and uniformly positive in the group of incomplete global ischemia/anoxia experiments. As in the complete models no consensus emerges as to best HBOT pressure, duration, frequency, number of treatments, or time to intervention. Shiokawa et al (1986) demonstrated an improvement in survival with 2 ATA HBOT for only 30 min, with best results at three hours post-insult as opposed to one hour. Weinstein et al (1986) achieved an 84% reduction in mortality with a 15 min 1.5 ATA treatment and Grigoryeva (1992) demonstrated a superior effect of 1.2 ATA/30 min over 2 ATA/60 min on survival and preservation of neuronal transcription. Yaxi et al ( 1995) and Yiqun et al (1995) both showed improvements in brain enzymatic function from a single HBOT treatment at 2.5 ATA with the Yaxi article suggesting a minimum 20-40 min duration of HBOT exposure for efficacy. Kondo et al (1996), meanwhile, showed that repetitive HBOT at 2 ATA/60 preserves hippocampal neurons and decreases heat shock proteins; there was a greater effect when the HBOT was started at six h instead of 24 h. Essentially, HBOT prevented delayed cell death (apoptosis) without oxygen toxicity. This anti-apoptotic effect was also proven by Calvert et al (2002). This as well as the Hai et al ( 2002) study used almost identical models to simulate human neonatal ischemia/hypoxia, a research and clinical subject of intense interest that has been heightened by review of the important neonatal resuscitation paper by Hutchison et al (1963) in the last edition of this textbook. In the Hai study repetitive 2.5 ATA HBOT delivered 7 d post insult had a signal induction effect on brain fibroblastic growth factor (bFGF) while also increasing the amount of bFGF; bFGF increase also occurred in the hyperbaric air group. Calvert showed that just a single 3.0 ATA HBOT preserves brain weight and minimizes apoptosis and these two effects translate to improved neurological function at five weeks of age. The only equivocal study, Mickel et al (1990), showed that normobaric oxygen (NBO) gave mixed results: increased production of white matter lesions while sparing cortical netirons. Lastly, the Wada et al ( 1996) study, the only model with pre-ischemic HBOT, proved an anti-apoptotic effect of HBOT. This is somewhat similar to the protective effect of pre-carbon monoxide exposure HBOT in the Thorn (1993a) model. Despite the inability to establish clear guidelines regarding HBOT parameters in incomplete global ischemia/anoxia, the results of HBOT treatment have been uniformly positive across the range of ischemic exposures (up to 3.5 h), with delays in starting HBOT treatments up to 7d, treatment pressures as low as 1.2 ATA and HBOT durations as short as 15 min. Importantly, almost all of the above studies initiated HBOT within three h of insult and showed positive results with a maximum of three treatments and in most cases only one. The three exceptions to this were the Kondo et al (1996) experiment which began HBOT at either 6 or 24 h (a 2-h group was used for a separate histological oxygen toxicity examination) after insult and continued treatment to a total of 14 or 35 treatments, the Wada et al (1996) experiment which used pre-ischemia/anoxia HBOT a maximum of 5 times, and the Hai et al (2002) study which delayed 10 daily HBOT’s for 7d. All of these studies were biochemical or pathological, not outcome/survival experiments. Interestingly, despite the prolonged course of HBOT exposure in Kondo’s study, no overt oxygen toxicity was delineated. The near uniform success of all of the above animal experiments suggests that pre-ischemic HBOT or single HBOT soon after ischemia is highly beneficial and probably the most important factor in positive outcomes while the absolute HBOT pressure is less important. The only exceptions to this conclusion are the Van Meter studies which strongly suggest that HBOT pressures of at least 2.8 ATA and ideally 4-6 ATA are necessary for cardiac arrest resuscitation.
Review of Human Clinical Studies
The human HBOT experience in cerebral ischemia/anoxia and coma is extensive, complicated, incomplete at times, and spread across multiple medical conditions. Despite the heterogeneous group of studies, the data shows a beneficial effect of HBOT, especially in the large series and particularly in traumatic brain injury (TBI).
Since the last edition of this chapter more reports have surfaced from the past 50 years and new data has accumulated, mostly in abstract form, for the hyperacute period. This data is consistent with previous reports and the abovementioned animal studies. In addition, more authors are recording improvements in the treatment of chronic brain injury, particularly in the pediatric population.
To facilitate review of the literature all reports have been categorized somewhat arbitrarily by amount of time delay to initiation of HBOT. Some reports span multiple categories and are unclear about exact times of HBOT intervention. In these a rough estimate was attempted based on the implications and inferences in the study, and references to companion articles. The four categories are hyperacute (≤ 3 h post insult), acute ( 4-48 h post), subacute ( 49 h to 1 month post), and chronic (greater than 1 month after insult).
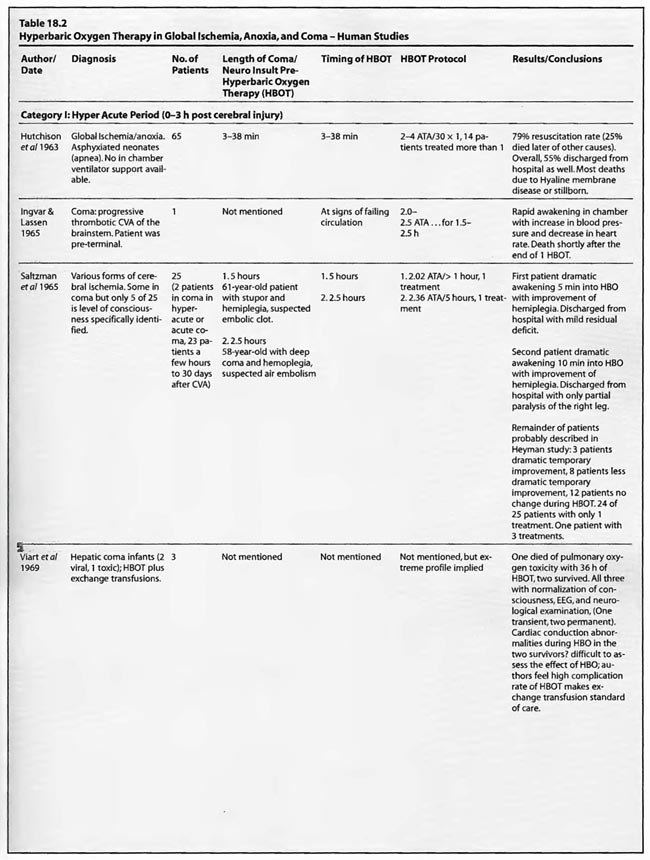
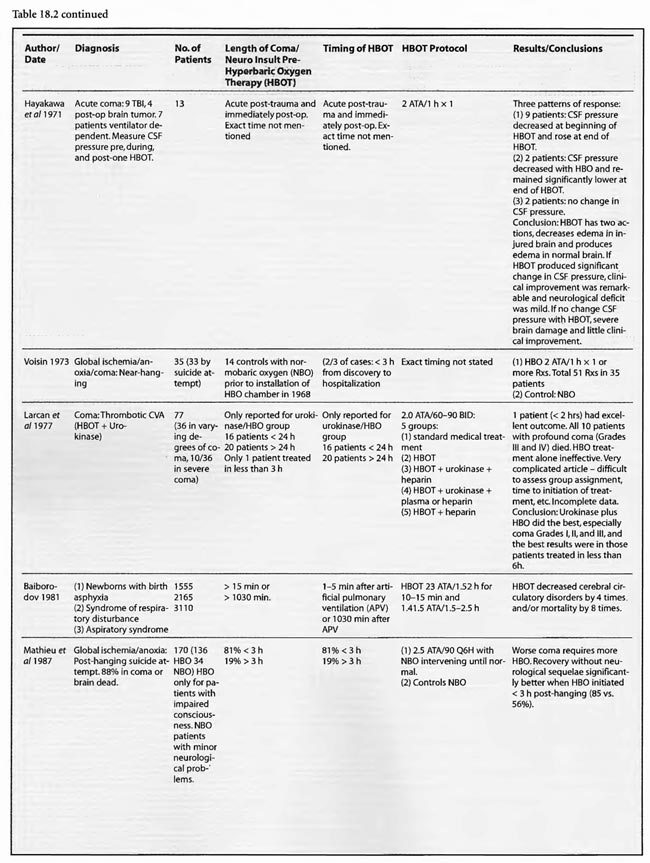
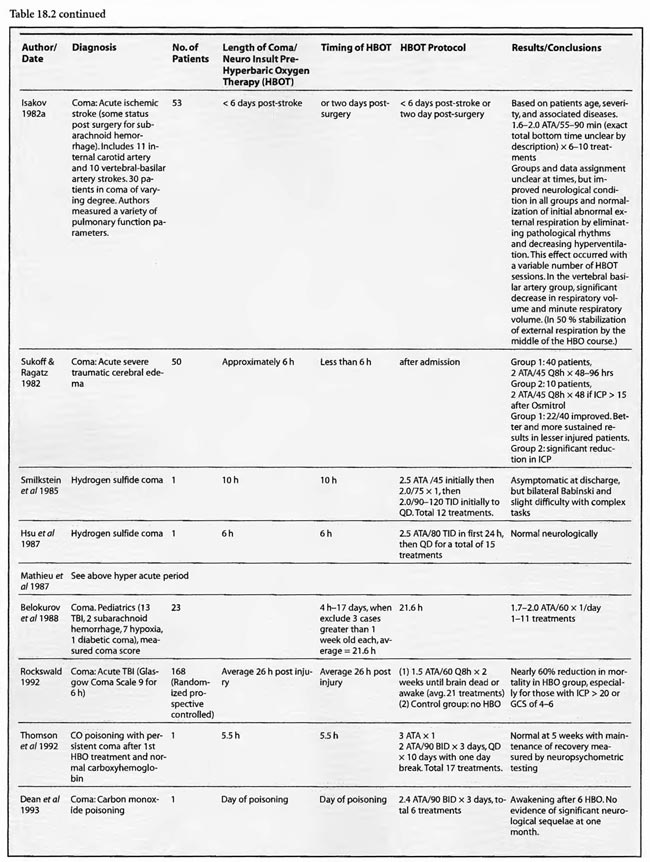
The studies of Mathieu et al (1987, 2000), Voisin (1973), Hutchison et al (1963), Kohshi et al (1993), Shn-Rong (1995), Larcan et al (1977), Viart et al (1969), Hayakawa et al (1971), Saltzman et al (1966), Ingvar and Lassen (1965), Baiborodov (1981), Sanchez et al (1999), and Van Meter et al (1999) address the hyperacute period of global ischemia/anoxia and coma. The most clear-cut of these are the studies reported by Mathieu et al (1987) and Hutchison et al (1963). Mathieu used HBOT in 170 cases of unsuccessful hanging, 34 of whom received NBO and 136 HBOT, and found statistically significant greater recovery without se quelae if HBOT was delivered < 3 hours post hanging (85 vs. 56%). Worse coma required more treatment, but even the worst only averaged 3.9 HBOT’s. The 34 NBO patients were those with minor neurological problems; only patients with impaired consciousness received HBOT. The pressure used in this study was 2.5 ATA/90 min. HBOT for near-hanging had become the standard of care at Mathieu’s facility in northern France following installation of the HBOT chamber in 1968 and the results of their historically controlled study of 1973 (Voisin). The study compared 14 patients with standard treatment (normobaric oxygen) pre-1968 to 35 patients with HBOT after 1968. HBOT reduced mortality by 39% and neurological sequelae by 59%. The authors stated that HBOT provided “a recovery both quicker and of better quality’: This series of 170 patients has been extended to 305 patients (Mathieu et al 2000) with nearly identical results, again strongly arguing for HBO-responsive pathology within the first few hours of rescue from near-hanging. These are similar to the initial results (79%) reported by Hutchison et al ( 1963) in resuscitation from neonatal asphyxia, but one-third of those patients regressed after the first treatment. Overall, 54% were discharged from the hospital as “well”. HBOT was used at 2-4 ATA in this study and treatment was initiated 2- 38 min after birth. These findings were replicated by Baiborodov (1981) at 2 to 3 ATA and 1.4- 1.5 ATA in 555 infants. Although the HBOT protocol is confusing and the details of the paper are limited due to publication of only the abstracts [the manuscripts were too numerous (350) and subsequently lost in a fire at one of the editors’ homes] the eightfold reduction in mortality is compelling and consistent with Hutchison’s data. More recently Sanchez et al (1999) has reported the same positive findings at 2 ATA in a small number of infants. Altogether these experiences with HBOT in neonatal ischemia/hypoxia/” asphyxia” are remarkable and significantly supported by the animal data, particularly the Calvert et al (2002) model.
Most of the other papers in this group imply hyperacute and acute treatment or do not state the time of HBOT intervention. Kohshi et al (1993) initiated HBOT soon after onset of symptomatic vasospasm in subarachnoid hemorrhage post-neurosurgical comatose patients using 2.5 ATA and an average 10 treatments which decreased subsequent progression to infarcts. Shn-Rong (1995) applied HBOT in 336 cases of cardiac arrest, near-drowning, unsuccessful hanging, electrocution, carbon monoxide, TBI, and other toxic and asphyxial coma patients, implying at least some treatment hyperacutely, with 75-100% recovery rates at 2- 2.5 ATA. Best results were with earlier treatment; delays to treatment required greater numbers of HBOT’s. In the Larcan et al (1977) paper it appears that only one patient was treated in the hyperacute period with both urokinase and HBOT. The result was excellent and the best in their study, but the effect can’t be necessarily attributed to HBOT alone. The Viart et al ( 1969) paper does not mention time to HBOT in the three cases of infant hepatic coma, but the profile of HBOT seemed extreme since one patient died of pulmonary oxygen toxicity with 36 h ofHBO and the other two experienced cardiac conduction abnormalities during HBOT, an extremely rare complication of HBOT. Despite the apparent complications, all three patients had normalization of consciousness, EEG, and neurological exam with HBOT, two permanently and one transiently. Hayakawa et al (1971) performed a single 2ATA/l h HBOT immediately after surgery on four comatose brain tumor patients and nine acute TBI patients. The time to initiation of HBOT was not stated but was probably< 3 hours in the post-operative patients. The authors found three patterns of CSF pressure and clinical response that corresponded to differential effects on normal and injured brain; HBOT decreases edema in injured brain and produces edema in normal brain. Most patients had an initial decrease of CSF pressure with HBOT and then return to pre-HBO level at the end ofHBO. The patients with a major decrease in CSF pressure during HBOT had remarkable clinical improvement and a mild neurological deficit If there was no change in CSF pressure the reverse was true. The duration of the HBOT neurological improvement was not mentioned.
The coma case reported by Ingvar and Lassen (1965) showed a transient rapid awakening of a patient with “failing circulation” but died at the conclusion of a 2-2.5 ATA/ 1.5-2.5 h HBOT. This could be the natural history of the patient’s disease and/or an oxygen toxicity effect. The other single case reports are of a “dramatic” near-complete cure of a suspected air embolism patient treated with a 2.36 ATA/5 h session of HBOT (Saltzman et al1966) and the Van Meter et al (1999) example of AGE, massive DCS, and cardiac arrest. The Saltzman treatment was similar to the United States Navy Treatment Table VI for air embolism and serious decompression sickness and may explain the near-complete cure without oxygen toxicity after a higher pressure very prolonged oxygen exposure (5 h). The Van Meter case also used a very prolonged deep oxygen exposure, 6 ATA pure oxygen on a modified US Navy TT6A extended to a US Navy TT7 with oxygen breathing periods at 3 ATA. This case spawned the guinea pig and swine resuscitation experiments of 1988 (Van Meter et al) and 1999/2001a,b (Van Meter et al), respectively. The success of the Saltzman and Van Meter cases are likely due to the large proportion of the pathology caused by intravascular gas. Overall, the preponderance of data in these 14 HBOT studies and 1,388 global ischemia/anoxia and coma patients is strongly positive with pressures greater than or equal to 2 ATA and a minimum of 1-7 treatments. The effect of lower pressures of HBOT can’t be stated since only one study reported pressures < 2 ATA (Baiborodov) and it is unclear if the lower pressure in this study was a tapered dose on the first HBOT or low pressure tailing treatments.
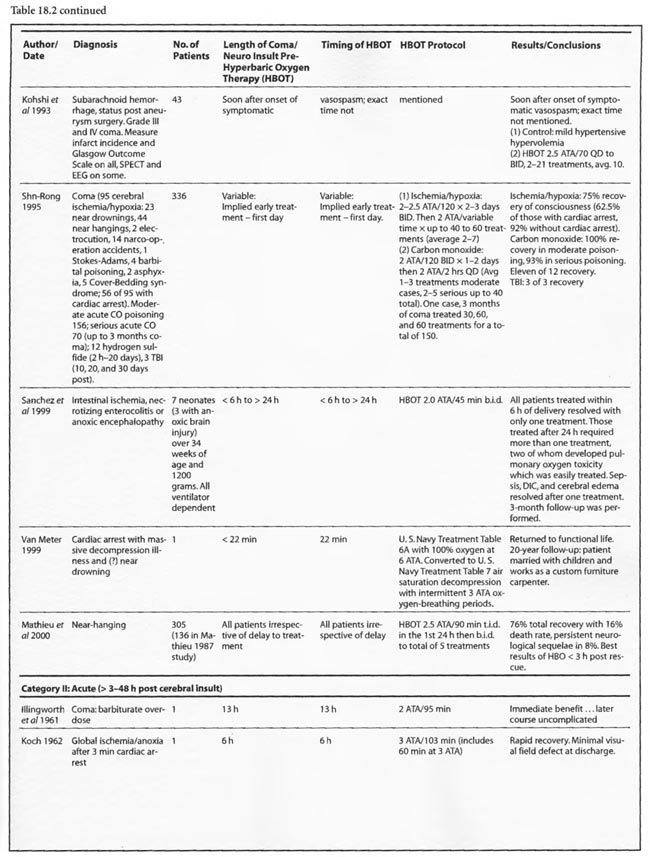
Thirty-one studies fall into the second or acute category. Once again the preponderance of data is positive, either transiently or permanently, regardless of the etiology of coma. Kohshi et al (1993), Mathieu et al (1987), Voisin (1973), Shn-Rong (1995),and Viart etal (1969) papers span this and the hyperacute period and were already reviewed. The Larcan et al (1977) study had one patient in the hyperacute period mentioned above and 35 coma patients in the acute period. HBOT appeared to have no effect and, in fact, was no different from the medical treatment group, but the data is incomplete, lacking a pure urokinase group and exact times to initiation of treatment. All ten severe coma patients died with lesser coma grades I- III showing the best response to combined urokinase plus HBOT, and minimization of time to treatment the best predictor of success. The Saltzman et al (1966) report also had one patient in both the hyperacute and acute periods. The acute patient had an embolic clot CVA and was treated 5 h post CVA at 2.02 ATA/> 1 h with near total permanent improvement. Lastly, nine TBI patients of Hayakawa’s et al (1971) 13 patients were most likely in the acute period, but the results have already been summed above.
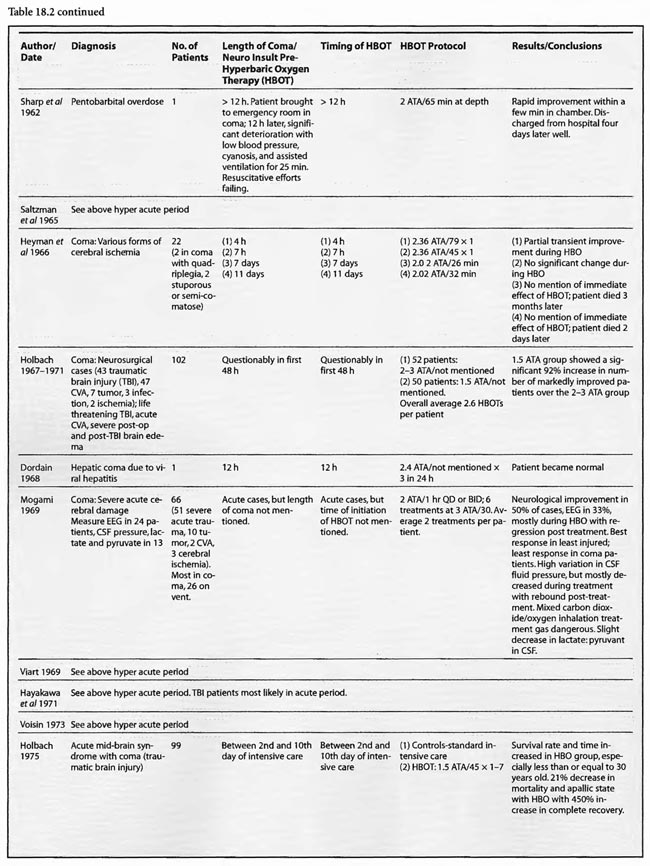
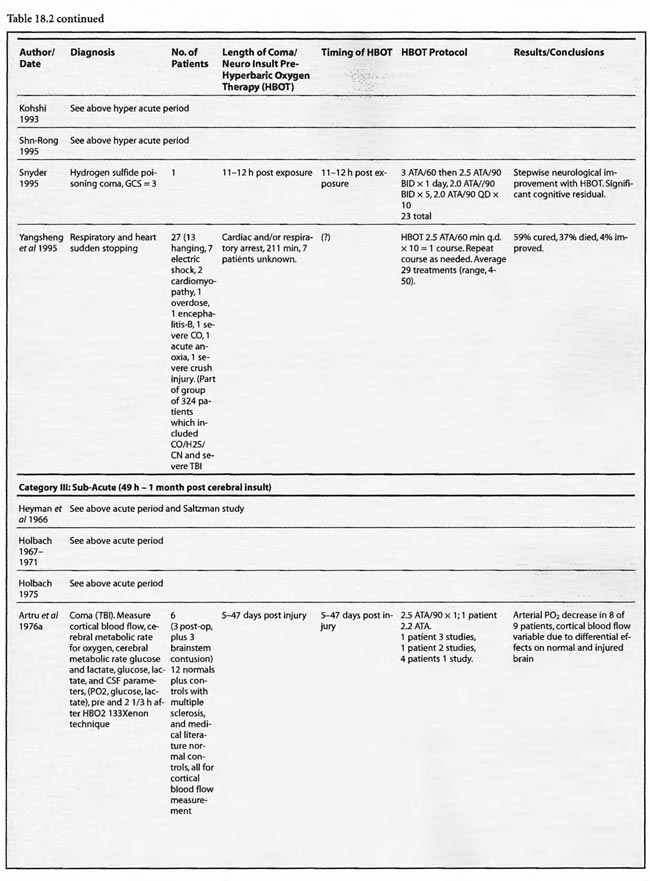
Of the remaining 24 studies, 15 were at pressures greater than or equal to 2 ATA: Sharp et al (1962), Heyman et al (1966), Mogami et al (1969), Sukoff (1982), Thomson et al (1992), Dean et al (1993), Snyder et al (1995), Yangcheng (1995), Dordain (1968), Illingworth et al (1961), Koch et al (1952), Hsu et al (1987), Smilkstein et al (1985), Sheffield et al (1976); 7 used 1.5 ATA:(Holbach a, b, d, e, f), Rockswald (1992), and Rockswold et al (2001); and two used 1.6-2 ATA: Belokurov et al (1988), Isakov et al (1982); with near uniform transient or permanent positive results. Four of the seven 1.5 ATA reports (Holbach et a/197 4a, 1977 d, e, f) compared 1.5 ATA to either 2, 2.5, or 2-3 ATA and demonstrated better results at 1.5 ATA, using a variety of clinical, biochemical, and physiological outcome measures. Four of the seven studies (Holbach et al 1974b, 1977d; Rockswald 1992, Rockswold et al 2001) initiated treatment > 24 h after injury and the fourth (Holbach et al 1974a) does not mention time to treatment, but implies treatment in the acute period since the patients are neurosurgical cases and they are reported incidentally in a paper on cerebral glucose metabolism in acute brain-injured patients which is a preliminary version of patients who were a “few days” post injury (Holbach 1977d). The fourth and fifth 1.5 ATA papers (Holbach 1977e, 1977f) span the acute and subacute periods and are similar patients to those in the other Holbach papers. In (Holbach 1974a) 1.5 ATA had significantly better clinical results than 2-3 ATA. This is confirmed with CBF measurements (Holbach 1977e), EEG (Holbach 1977f), and cerebral glucose metabolism (Holbach 1977d) where 15- 30 min excursions to 2 and 2.5 ATA caused deterioration in the measured parameters. While the Holbach (1977d) experiment did not explore pressures between 1.5 and 2 ATA, the Belokurov study affirmed the efficacy of 1. 7-2 ATA pressures in 23 comatose children with 100% recovery of consciousness. Their results were maximal in TBI and if initiated < 24 h post coma. Similarly, Isakov et al (1982) experienced good results between 1.6 and 2 ATA in patients with cerebrovascular accidents. The final study (Rockswold et al 2001) deserves special comment. This was an elegant follow-up study to Rockswald (1992) to evaluate the cerebral and biochemical physiological effects of 1.5 ATA HBOT on acute severe TBI. The authors demonstrated that 30 min total dive time had achieved the maximum reduction of elevated ICP in chamber, one HBOT recoupled flow/metabolism in injured brain and reduced lactate levels, and the HBOT changes persisted at least six h after HBOT. The importance of the study is its duplication of the Holbach studies’ elucidation of HBOT effects on pathophysiology in acute severe TBI and the underpinning of all of the clinical studies, most notably the Rockswald 1992 study with its HBOT induced 47- 59% reduction in mortality. The study emphatically argues for HBOT in acute severe TBI. 518 of the 705 patients reviewed in this category (excluding Kohshi, Mathieu, Voisin, Shn-rong, and Viart) had TBI; the data strongly argues for the routine use of HBOT in TBI at 1.5 ATA. Overall, treatment courses tended to be longer in the acute category than the hyperacute, using higher HBOT pressures earlier (less than or equal to 24 h) and lower pressures later, with overall positive effects regardless of coma etiology: chemical/toxic gas, trauma, CVA, surgery, etc.
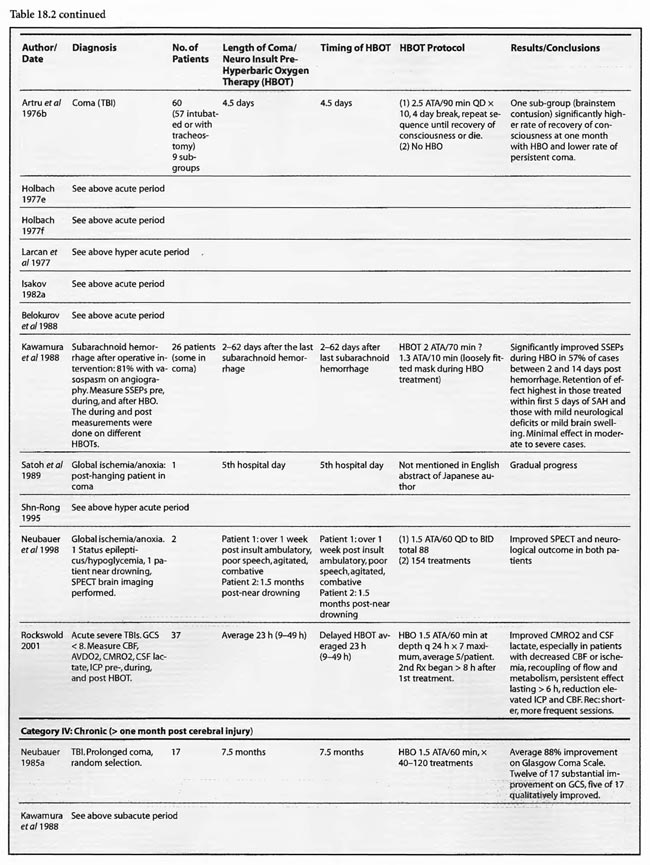
In the third category, subacute ( 49 h-1 month) sixteen studies are presented. The papers of Holbach (1974a,b, 1977e,f), Larcan et al (1977), Shn-Rong (1995), Isakov et al (1982), and Belokurov (1988) were discussed above, but to reiterate, many of the TBI cases of Holbach started HBOT 2- 10 days post injury. Results were positive and favored treatment at 1.5 ATA for 1-7 times. Lareng et al (1973) reported two additional late TBI coma cases and had excellent outcomes with prolonged treatment at 2.0 ATA. In the Shn-Rong series a number of carbon monoxide cases presented with > 6 days of coma. In general they required more treatment, and one case of 90 day coma was cured finally with normal EEG after 150 treatments in three stages. Three patients with TBI coma of 10, 20, and 30 days regained consciousness after 7- 20 treatments. Almost all except the initial few treatments were at 2 ATA. Two additional TBI studies by Artru et al (1976a, 1976b) had mixed results. The first involved 60 TBI patients 4.5 days post injury, HBOT at 2.5 ATA, and an average 10 treatments with multiple breaks in protocol, and few receiving much treatment in the first week. Only one of nine sub-groups (brain stem contusion) achieved significant improvement with HBOT (see above discussion on penumbra/umbra size considerations in brainstem vs. cortical coma). The second study with 6 patients, 5-47 days post insult, examined blood flow, metabolism, and CSF biochemistry before and after 2.5 ATA HBOT. Results were inconclusive, but arterial partial pressure of oxygen declined in 8 of 9 patients, CSF oxygen remained elevated above baseline for 2 h after HBOT, and the authors concluded that HBOT has different effects on normal and injured brain circulation. Both of these studies featured high pressure, 2.5 ATA, later in the course of illness and are consistent with an oxygen toxicity effect as Holbach demonstrated in multiple reports above. The final five studies deal with subacute CVA (Holbach 1976c), (Heyman et al1966), subarachnoid hemorrhage with postoperative vasospasm (Kawamura et al1988) , post-hanging (Satoh et al 1989), and status epilepticus/hypoglycemia (Neubauer et al 1998). Holbach reported excellent results at 1.5 ATA, Heyman did not mention immediate effects at 2.02 ATA, Kawamura noted sustained improvement in SSEP’s at 2 ATA, Satoh noted “gradual progress” of his unsuccessful hanging patient and Neubauer found significant progress at 1.5 ATA. In summary, with delay to treatment of 2-30 days generally positive results are achieved with HBOT with a tendency to lower pressures and longer treatment courses.
The final category, chronic (> 1 month), has seen a substantial increase in studies since the previous edition. There are now 16 studies, most of which address pediatric brain injury. Six of these are single cases and the remainders are prospective and retrospective case series and prospective controlled trials. All of the reports used 1.5-2.0 ATA oxygen with most < 1.75 ATA, except for Golden et al (2002) and Miura et al (2002), which used 1.25-2.5 ATA and 2.0 ATA, respectively. Treatment times were mostly < 60 min at depth and extended from 1 to over 500 treatments; most involved 20 to 40 treatments. The 2nd International Symposium studies were grouped together due to their heterogeneity of subjects, protocols, and hypotheses. For example, the Harch (2001) article addressed and identified oxygen toxicity and/or negative side effects of lower pressure HBOT in chronic brain-injured patients with prolonged treatment courses at 1.5 ATA (average 119 treatments) and 1.75 ATA (91 treatments) or early in treatment at 1.75 ATA or greater. In addition, a withdrawal syndrome (4 patients) was described in brain-injured individuals habituated to 1.75 ATA HBOT. In two of these four cases the author intervened and truncated the neurological deterioration with additional HBOT at a lower pressure. These toxicity findings were reaffirmed by Miura et al (2002) in a case of delayed neuropsychiatric sequelae from drug overdose complicated by hypoxia. The patient recovered acutely then deteriorated to akinetic mutism. The authors initiated HBOT at 2.0 ATA/90 min and the patient improved then worsened during prolonged HBOT, identical to cases in Harch (2001). The behavioral problems slowly abated upon cessation of HBOT, but his cognitive decline continued. The case suggests over-treatment with HBOT resulting in transient and permanent negative side-effects against a background of permanent clinical improvement. MRI FLAIR, MRS, and EEG tracked the clinical course in a manner that was almost identically to the SPECT findings in the cases of Harch (2001).
In conclusion, additional experience with HBOT in chronic global ischemia, anoxia and coma that was supported by SPECT results has accumulated in the past five years, strongly suggesting a positive trophic effect, but the controlled studies have confused the scientific community due to flaws in the design and methods of these studies.
Case Studies
To illustrate the effect of HBOT in both acute and chronic global cerebral ischemia/anoxia and coma several cases treated by these authors are presented below. In each case the visual medium of SPECT brain blood flow imaging on a high resolution scanner (7 mm; Picker Prism 3000) registers in a global fashion the neurocognitive clinical improvement experienced by the patients and witnessed by the authors. The SPECT brain scans presented below are CT technology with the patient’s left brain on the reader’s right and vice versa, with the 30 frame images registering transverse slices from the top of the brain in the left upper corner to the base of the brain in the lower right corner. Images are approximately 4 mm thick. Brain blood flow is color coded from white-yellow to yellow to orange to purple, blue and black from highest brain blood flow to lowest. Normal human brain shows predominantly yellows and oranges, but, more importantly, has a fairly smooth, homogeneous appearance. The companion image (B) to the 30 slice transverse set of images is a three-dimensional surface reconstruction of the transverse images. Abnormalities in perfusion are registered as defects and as coarseness of the brain’s surface.
Patient 1: HBOT Treatment for Coma Due to Traumatic Brain Injury
A 19-year-old male was inadvertently ejected from a motor vehicle at 65 mph with impact on the left frontal/parietal region of the skull. Within one-half hour Glascow coma scale was 6- 7 and the patient was ventilator dependent. CT of the brain revealed diffuse edema, midline shift, petechial hemorrhages, subarachnoid hemorrhage, small subdural hematoma, and basilar skull fracture. HBOT was given 19 h post injury at 1.75 ATA/90 bid. On the first treatment the patient began to fight the ventilator. Initial SPECT brain imaging obtained five days post injury on a single-head low-resolution scanner was “normal.” Repeat SPECT imaging on a triple-head high –resolution scanner occurred 30 days post injury (Figure 18.1, A and B) and now clearly demonstrated the significant injury to the left frontal area as well as the contra coup injury to the right parietal/occipital area characterized by luxury perfusion. Nine days later and two hours after a fifth additional HBOT, SPECT was repeated (Figure 18.2, A and B) and showed a dramatic “filling in” of the injured areas thus giving functional neurophysiological support to the clinical decision to continue HBOT. The patient, meanwhile, progressed rapidly on twice daily HBOT for four weeks with often new neurological or cognitive findings occurring in the chamber and then continued on HBOT 4 times a day for seven weeks, at which time he was conversant and independently ambulatory with slight spasticity. At 11 weeks the patient was transferred to a rehabilitation center and his HBOT discontinued by the new medical team. SPECT imaging at this time (Figure 18.3, A, B, and C) registers the patient’s clinical progress with a persistent increase in flow to the left frontal region while some deterioration occurs to the area of previous luxury perfusion on the posterior right. The patient made transient limited initial progress at the rehabilitation center then quickly leveled off cognitively while his spasticity and balance worsened. Three months after discontinuance of HBOT the patient’s father requested further HBOT and repeat SPECT brain scan (Figure 18.4, A and B), psychometric, and motor testing were obtained. SPECT now demonstrates a significant deterioration in the right frontal and posterior areas, while the left frontal normalization persists. The right posterior area has infarcted on simultaneous MRI. To assess recoverable brain tissue the patient underwent a single 1.75 ATA/90 min HBOT followed by SPECT imaging (Figure 18.5, A and B); SPECT showed improvement in the right frontal and parietal/occipital lesions along the ischemic penumbral margins. HBOT was resumed for an additional 80 treatments, once/day at 1.75 ATA/ 90 min. The patient made a noticeable improvement in cognition ( 40 percentile gain in written computational mathematics), insight (the patient now verbalized for the first time the understanding that he had sustained a brain injury and could no longer aspire to be a surgeon), and balance (improvement in gait and progression from a 3-wheel tricycle to a 2-wheel bicycle). HBOT ( 188 treatments total) was discontinued when the patient desired enrollment in remedial courses at a community college. SPECT imaging at this time (Figure 18.6, A and B) shows improvement in perfusion in the ischemic penumbral areas of the right-sided lesions. The left hemisphere remains intact. In summary, HBOT, when reinstituted following SPECT and relapse after discontinuation of HBOT, prevented further deterioration and improved SPECT image as well as neurocognitive function in TBI, demonstrating the benefit of HBOT in the chronic stage of TBI.
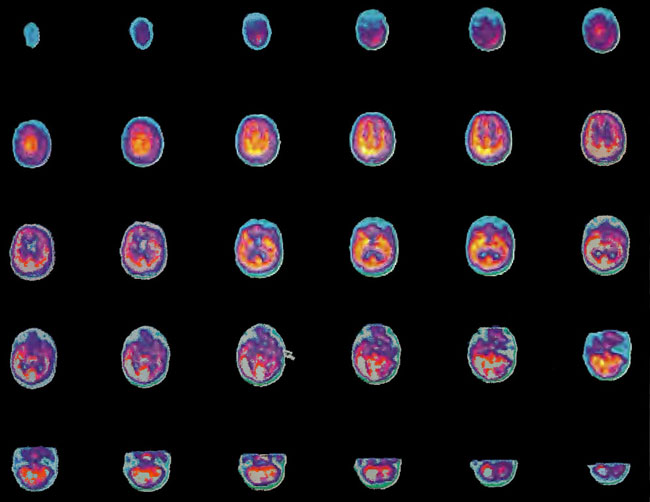
Figure 18.1A. HMPAO SPECT brain imaging, transverse slices, one month post injury. Note severe reduction in left frontal, parietal, and temporal brain blood flow with luxury perfusion in the right occipital parietal region.
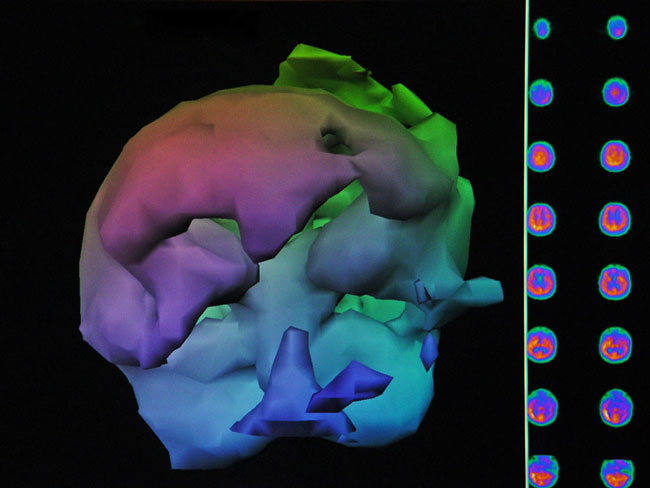
Figure18.1B. Frontal projection three-dimensional surface reconstruction of Figure 18.1A. Non cerebral uptake is shown in scalp and neck soft tissues.
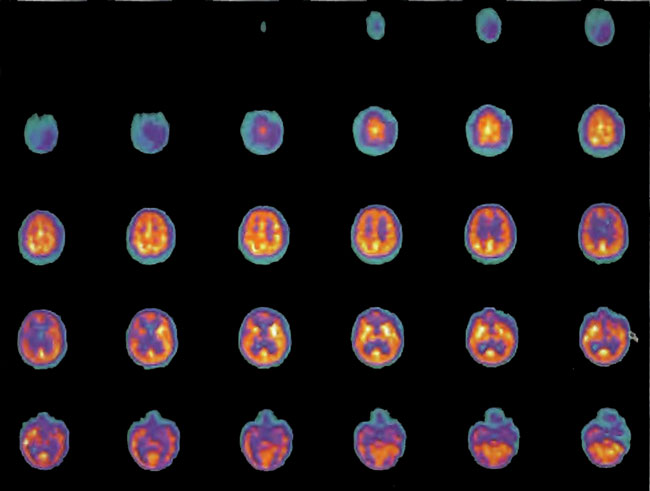
Figure 18.2A. HMPAO SPECT brain imaging, transverse slices, 9 days after Figure lS.lA and lS.lB and 2 hours post 5th additional hyperbaric treatment. Note improvement in flow to the left frontal, parietal, and temporal regions while defects begin to appear in the right frontal and parietal area. Luxury perfusion is no longer evident.
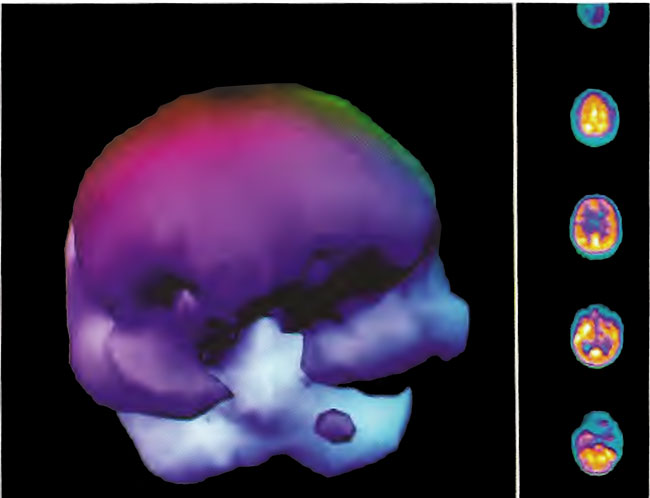
Figure 18.2B. Frontal projection three-dimensional surface reconstruction of Figure l8.2A.
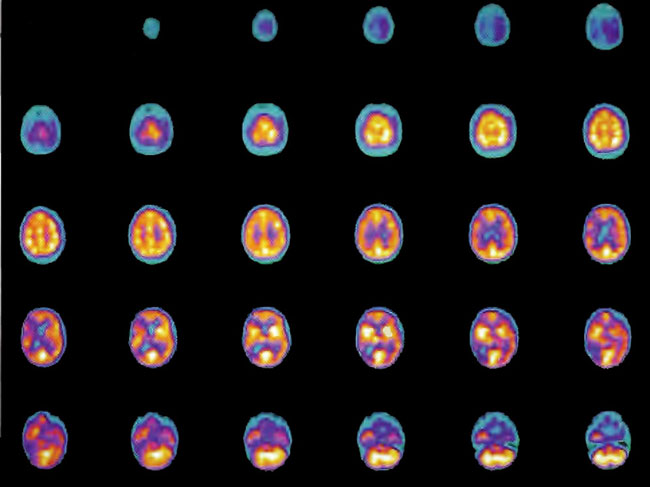
Figure 1 8.3A. HMPAO SPECT brain imaging, transverse slices, 11 weeks and 108 hyperbaric treatments post injury. Note maintenance of perfusion in the left frontal, parietal, and temporal regions with further progression of defects in the right frontal-parietal and posterior parietal-occipital areas.
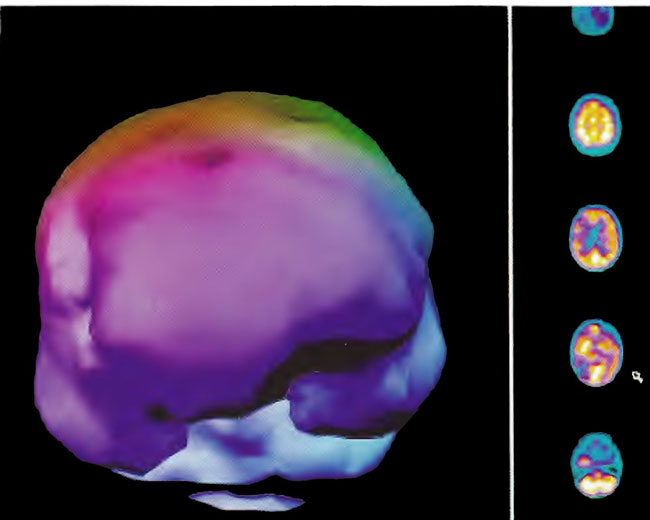
Figure 18.3B. Frontal projection three-dimensional surface reconstruction of Figure 18.3A.
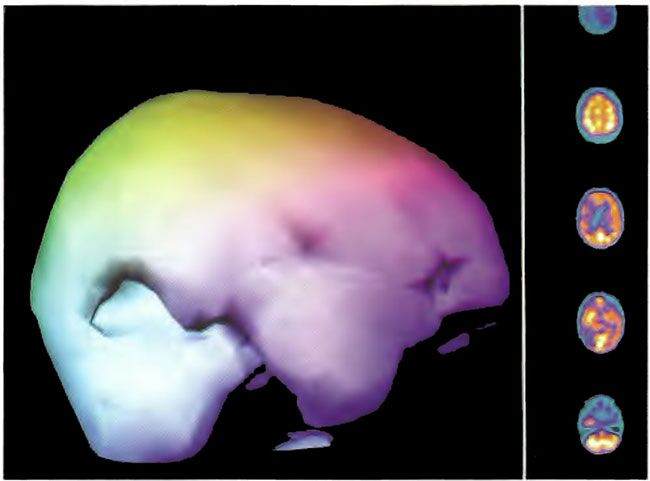
Figure 18.3C. Right lateral projection three-dimensional surface reconstruction of Figure l8.3A.
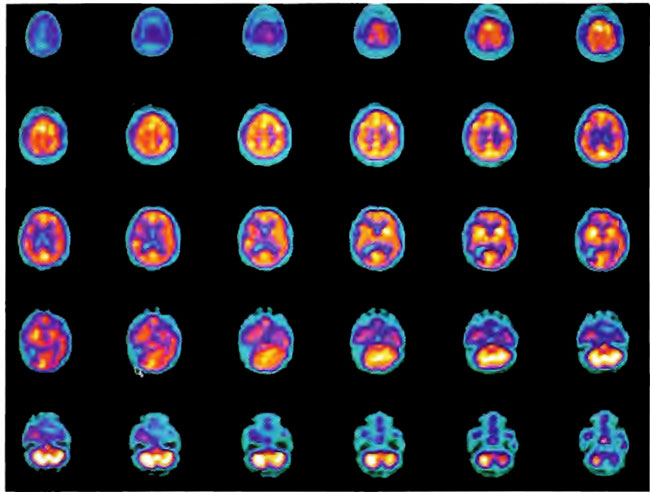
Figure 18.4A. HMPAO SPECT brain imaging, transverse slices, 3 months after Figures l8.3A, 18.3B, and l8.3C. Left frontal, parietal, and temporal perfusion is maintained with further deterioration of the right frontal and posterior defects
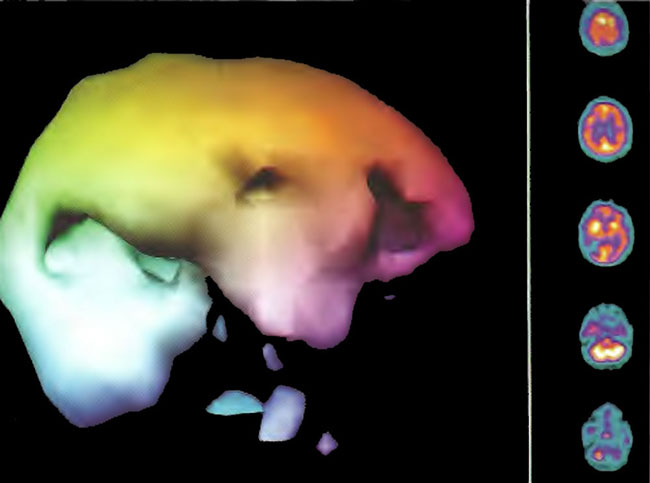
Figure 18.4B. Right lateral projection three-dimensional surface reconstruction of Figure 18.4A.
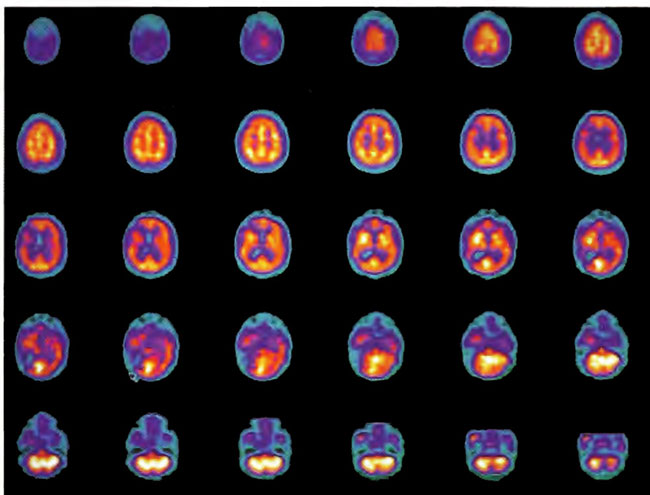
Figure 18.5A. HMPAO SPECT brain imaging, transverse slices, 2 hours following single HBOT at 1.75 ATA/90min. Note improvement in the right frontal and posterior defects.
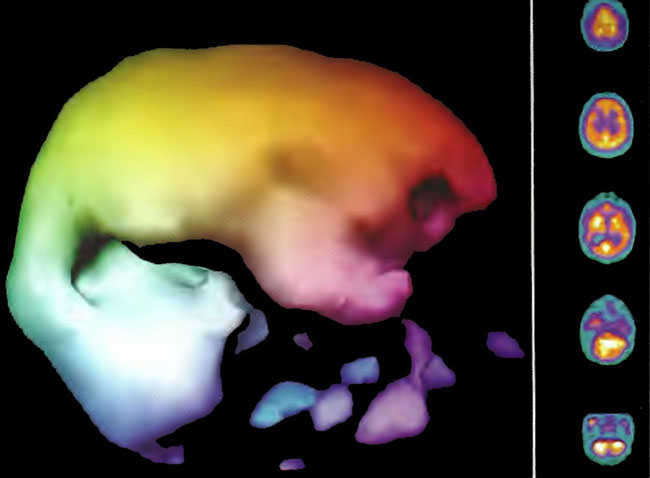
Figure 18.5B. Right lateral projection three dimensional surface reconstruction of Figure l8.5A.
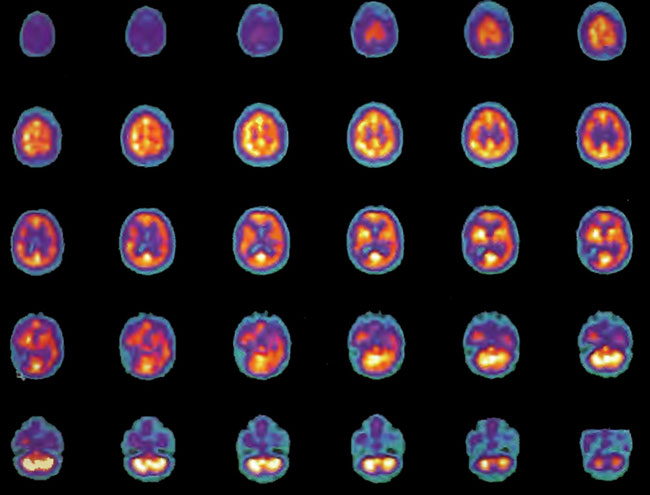
Figure 18.6A. HMPAO SPECT brain imaging, transverse slices, 5 months and 80 HBOT’s after Figure l8.4A. Note improvement in flow to the ischemic margins of the right frontal and posterior defects.
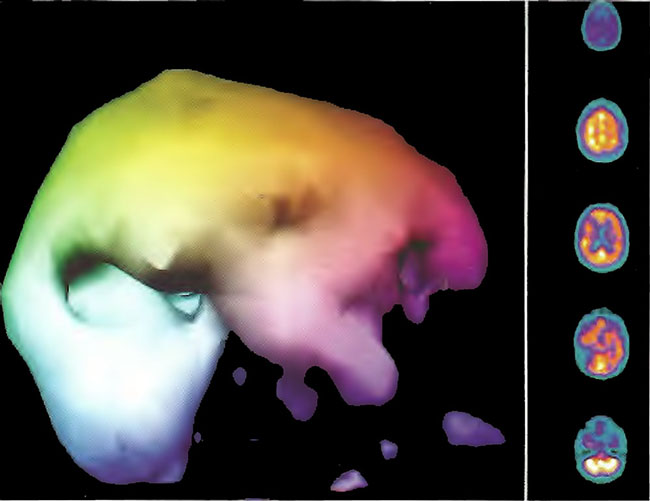
Figure 18.6B. Right lateral projection three dimensional surface reconstruction of Figure18.6A.
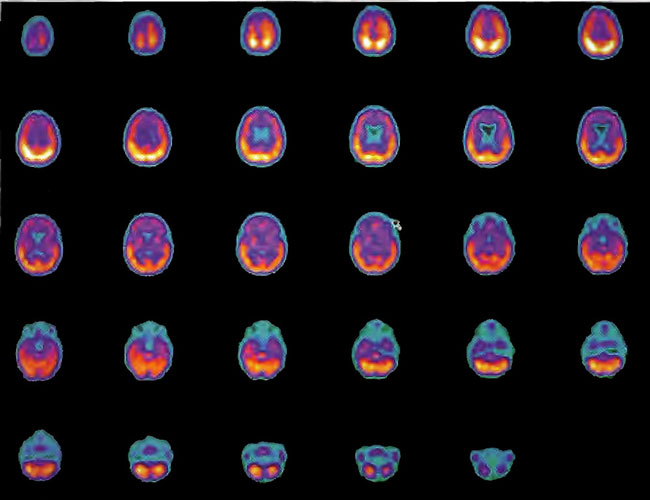
Figure 18.7A. HMPAO SPECT brain imaging transverse slices, baseline study two years status post near drowning. Note considerable reduction in frontal blood flow.
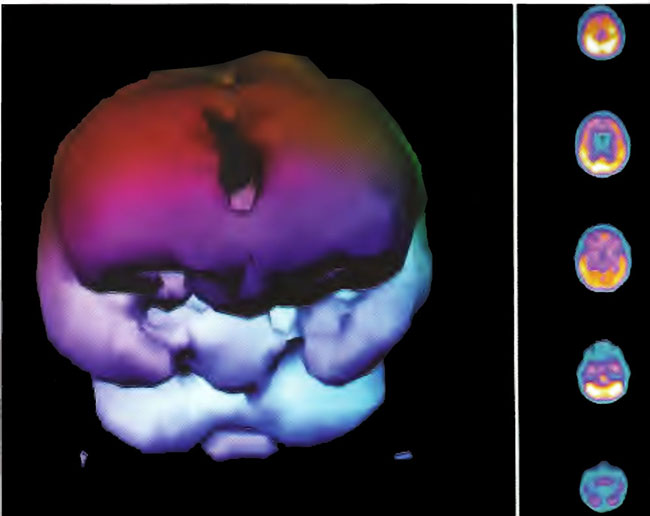
Figure 18.7B. Frontal projection three dimensional surface reconstruction of Figure 18.7 A.
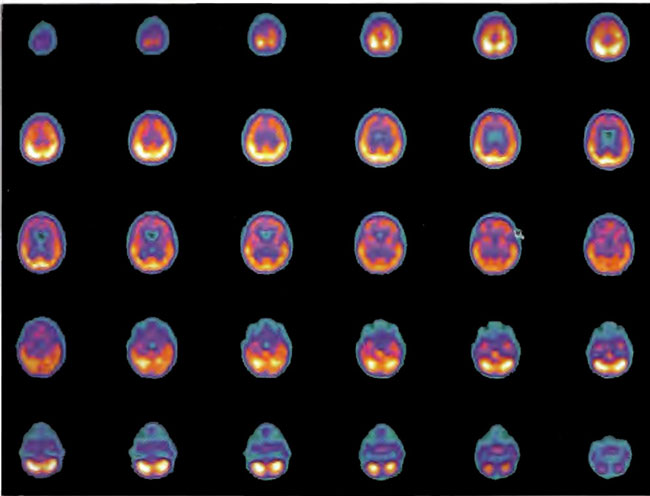
Figure 18.8A. HMPAO SPECT brain imaging, transverse slices, 1 day after Figure 18.8A and 2 hours following single HBOT at 1.5 ATA/60 min. Note diffuse increase in perfusion to the frontal lobes and improvement in overall brain blood flow.
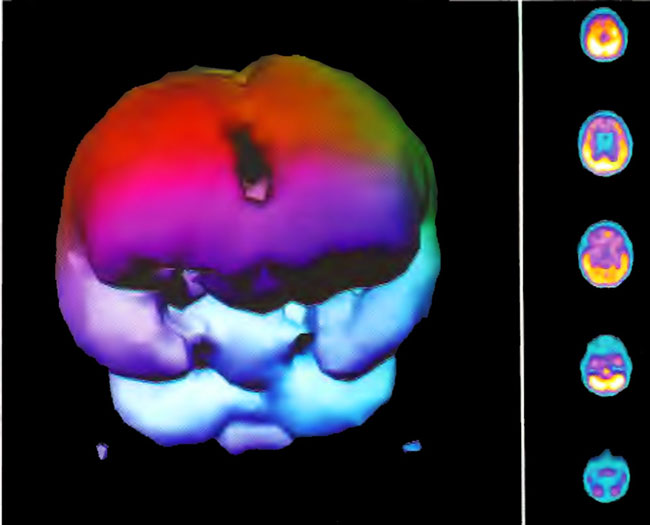
Figure 18.8B. Frontal projection three dimensional surface reconstruction of Figure 18.8A.
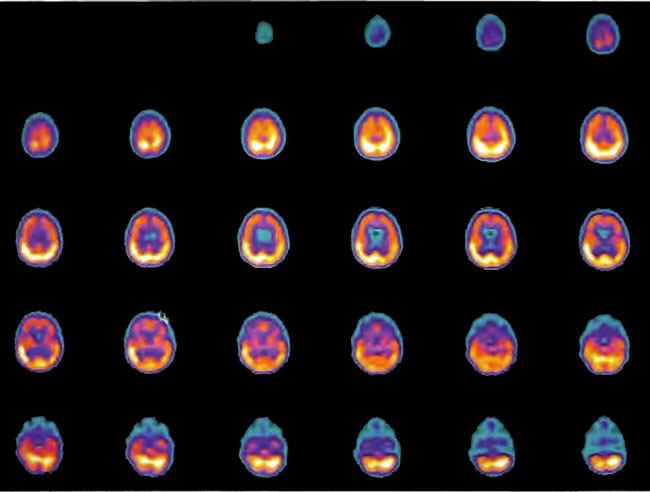
Figure 18.9A. HMPAO SPECT brain imaging, transverse slices, 4 months and 80 HBOT’s following Figure 18.9A. Note persistent increase in perfusion to the frontal lobes.
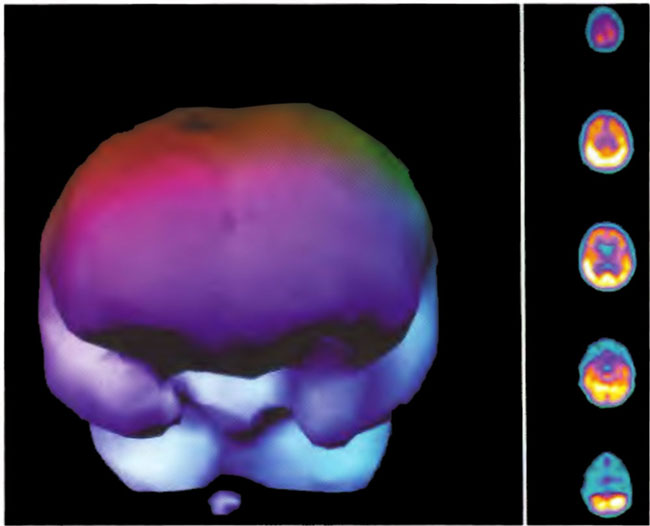
Figure 18.9B. Frontal projection three dimensional surface reconstruction of Figure l8.9A.
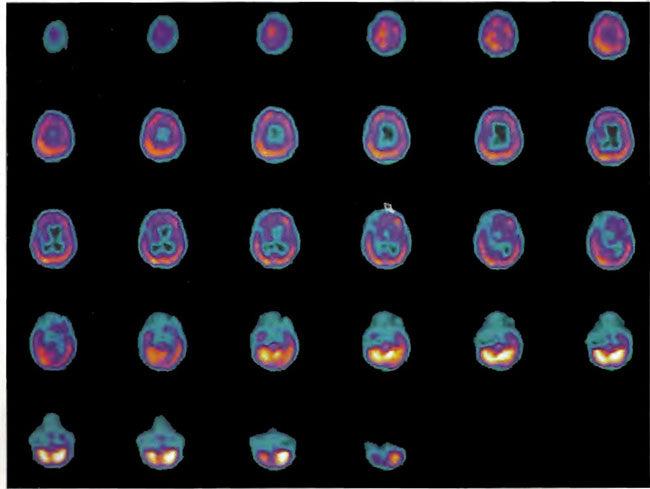
Figure 18.10. HMPAO SPECT brain imaging, transverse slices, baseline study two years post near drowning.
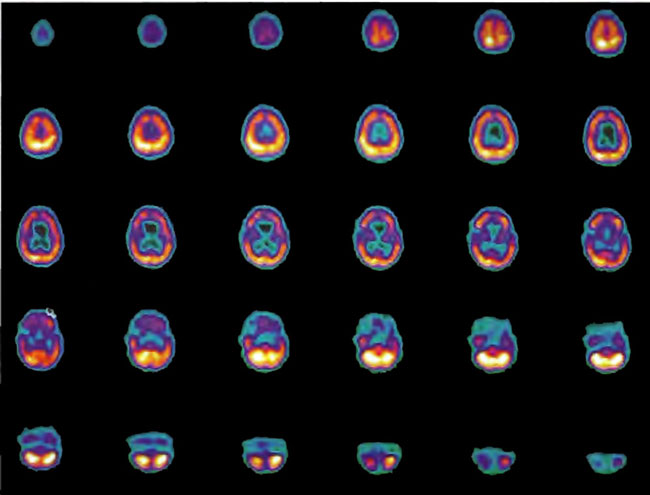
Figure 18.11. HMPAO SPECT brain imaging transverse slices one day after Figure 18.10 and two hours after a single HBOT treatment at 1.5 ATA/60min. Note generalized improvement to frontal lobe brain blood flow.
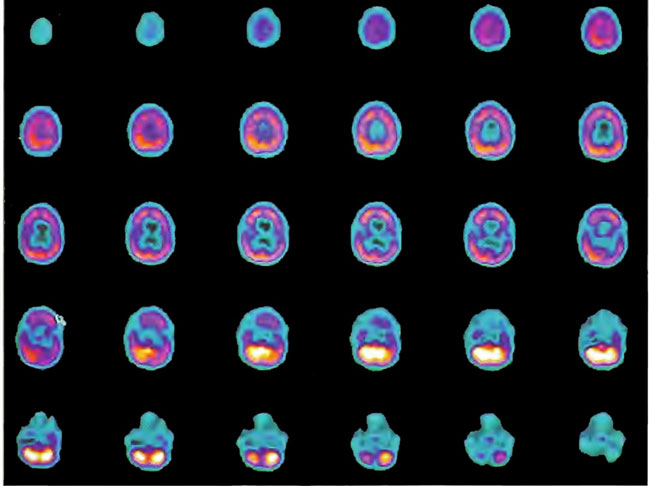
Figure 18.12. HMPAO SPECT brain imaging transverse slices four months and 80 HBOT treatments after Figure 18.10. Note persistent improvement to blood flow to the frontal lobes over baseline scan of Figure 18.10A.

Figure 18.13. ECD SPECT brain imaging transverse slices. Baseline study is on the right and after 38 HBOT treatments on the left. Note the predominant thalamic, midbrain, brainstem, and posterior fossa flow and lack of cortical flow on the first study which is augmented by cortical flow on the after HBOT study.
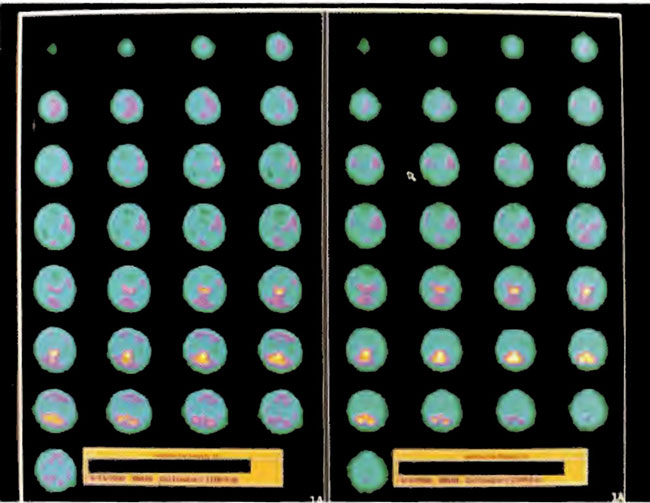
Figure 18.14. ECD SPECT brain imaging transverse slices. Baseline study is on the right again and study after 80 HBOT treatments is on the left. Note the improvement in cortical blood flow after HBOT.
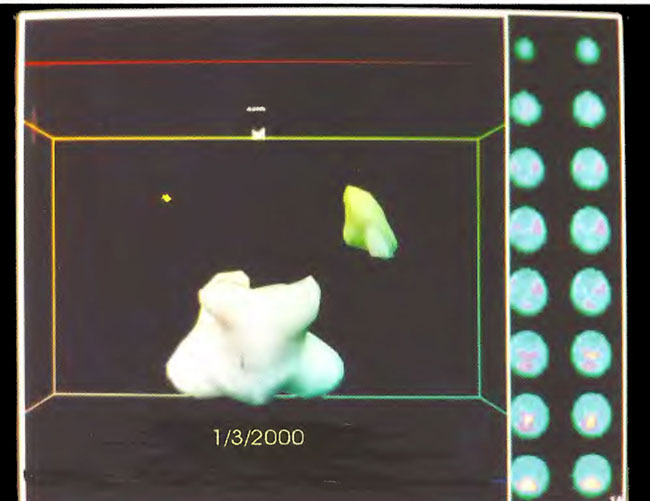
Figure 18.15. Frontal projection three-dimensional surface reconstruction of baseline study in Figure 18.13. Note the relative absence of cortical and striatal flow.

Figure 18.16. Frontal projection three-dimensional surface reconstruction of second study (after 38 HBOT treatments) in Figure 18.13. The patient has developed some cortical blood flow.
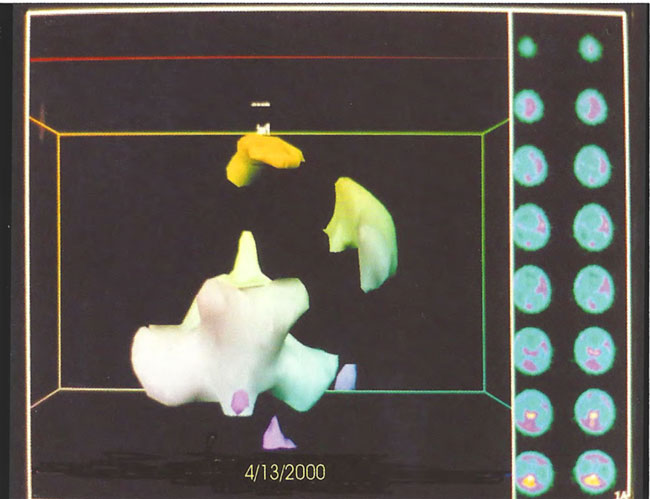
Figure 18.17. Frontal projection three-dimensional surface reconstruction of third study (after 80 HBOT treatments) in Figure 18.14. Note persistence of cortical blood flow.
Patient 2: Near Drowning, Chronic Phase
The patient is a 4-year-old male who was found at the bottom of a swimming pool after an estimated 5 min of submersion. Resuscitation measures were instituted and a pulse was regained 45 min after removal from the pool. Two years after the injury, the patient was wheelchair bound with significant motor disabilities, inability to speak and communicate, and problems with drooling, attention span, and swallowing. SPECT brain imaging was performed on a high resolution scanner before (Figure 18.7, A and B) and two hours after (Figure 18.8, A and B) a 1.5 ATA/60 min HBOT. The baseline scan in Figure 18.7 A shows a severe reduction in blood flow to the frontal lobes, while Figure 18.8A shows a generalized improvement in brain blood flow, particularly to the frontal lobes, and denotes recoverable brain tissue after the single hyperbaric treatment. The patient embarked on a course of 80 hyperbaric treatments at 1.5 ATA/60 min four times a day, 5 days per week with a 3 week break at the 40 treat ment point. At the end of 80 treatments, he returned for evaluation and was noted to have a generalized improvement in spasticity, movement of all 4 extremities, increase in trunk and head control as well as improvements in swallowing, awareness, non-verbal communication, and attention span. There was a global increase in blood flow on SPECT brain imaging performed at that time (Figure 18.9, A and B).
Patient 3: Near Drowning, Chronic Phase
Case 3: The patient is a 4-year-old boy who is 2 years status post 30 min submersion in a pond. Resuscitation regained a pulse 45 min after removal from the water. Two years later, the child is severely disabled with almost no cognition, frequent posturing, inconsistent tracking, extreme difficulty swallowing fluids, choking, and 10 petit mal seizures a day. Baseline SPECT brain imaging is shown in Figure 18.10 with prominent abnormalities in the inferior frontal lobes. The patient underwent a single HBOT at 1.5 ATA/60 min with repeat SPECT imaging 2 h after chamber exit (Figure 18.11). A generalized improvement in flow is noted, particularly to the frontal lobes, identifying potentially recoverable brain tissue. The patient underwent a course of 80 hyperbaric oxygen treatments at 1.5 ATA/ 60 min QD, 5 days per week with approximately one month break after 40 treatments. On return evaluation, SPECT brain imaging was repeated (Figure 18.12) . Improvement in frontal lobe blood flow is noted over the baseline scan. The child exhibited greater awareness, control of his head, eye tracking, alertness, nonverbal communication, and performance of some simple commands, improvement in swallowing and decrease in seizure frequency.
Patient 4: Battered Child Syndrome
Case 4: The patient is a 6-month-old girl who was slammed against the mattress of her crib by her father on multiple occasions over a four day period at two months of age. One of the first episodes was characterized by a short period of apnea; paramedics arrived at the house and found the child to be apparently normal. Three days later another episode of shaking ended with deliberate suffocation and cardiac arrest. Resuscitation was complicated by multiple recurrent arrests en route to and at the hospital. CT of the brain revealed bilateral subdural hematomas and subarachnoid hemorrhage and CT of the cord, Ll to L4 subdural hematoma. The child was ventilator dependent for 12 days. Seizures developed. Repeat CT 18 days after arrest showed severe diffuse encephalomalacia, bilateral infarcts, and bilateral hemorrhages with sparing of the basal ganglia, posterior fossa, and brainstem. EEG showed seizure activity on a background of minimal electrical activity. The mother unsuccessfully sought HBOT therapy. Four months after the injury the child was stable enough to travel to New Orleans where she was found to be paraplegic with rectal prolapse secondary to loss of sphincter tone. She was unable to suck and was dependent on a feeding tube. She had 5-8 seizures/day and was unable to interact socially. The patient received 38 HBOT sessions at 1.5 ATA/60 min TDT, Sd/wk with progressive neurological improvement. She was more awake and aware, starting to interact with her mother, had better head control, use of her arms, and no seizures. SPECT brain imaging reflects this improvement in Figure 18.13; baseline scan is on the right and one after 38 HBOT’s is on the left. There is a remarkable diffuse increase in cortical blood flow after HBOT with a relative absence of flow on the baseline scan, consistent with the EEG. The day after her 38th HBOT treatment the patient began a four week phenobarbital taper. HBOT was re-instituted at l.5 ATA/60 qd, six treatments in Sd/week for 42 more treatments (total of 80 HBOT treatments) 2 weeks into the taper. By the 80th HBOT treatment, the patient had begun a phenytoin taper, was eating baby food, had been weaned off Propulsid for her reflux disorder, was much more alert, had increased motor activity, truncal balance, improved social interaction/early smile, much less irritability, a return of rectal tone, and resolution of rectal prolapse, but was still paraplegic. Repeat SPECT brain imaging after 80 HBOT treatments again captures this increased clinical activity with increased cortical blood flow in Figure 18.14 (baseline scan is again on the right and after 80 HBOT treatments on the left). Three dimensional surface reconstructions of the three scans are shown in chronological order in Figures 18.15, 18.16, and 18.17. Repeat EEGs after 65 HBOT treatments (patient off phenobarbital, now only on phenytoin) and one month after her 80th HBOT treatment (off all anticonvulsants) showed no seizure activity. EEG also showed new background rhythm and bursts of frontal activity.
Conclusions
There are several causes of coma and global cerebral ischemia/anoxia. HBOT has been used in a variety of animal models and in over 2500 patients with these conditions worldwide. Although HBOT protocols have varied, the results have been remarkably consistently positive with improvement in a variety of physiological and biochemical measures and outcomes, the most important of which was improvement in overall clinical condition and consciousness. This consistent success rate suggests a generic effect of HBOT on common brain pathophysiological processes at different stages in global ischemia/anoxia and coma. Importantly, his review excluded thousands of cases of acute carbon monoxide (CO) coma in the medical literature treated with HBOT because of the confusion over HBOT effects on the metabolic poison and COHb dissociation vs. hypoxia and other pathophysiology. No doubt hypoxia is a major contributing insult to the patient’s overall condition in CO and reperfusion injury a significant component of the pathophysiology, and both of these are treated definitively by HBOT early after extrication, but many patients arrive for HBOT hours after extrication (Thorn 1992; Goulon et al1969; Raphael et al1993), adequately oxygenated, with low or normal COHb levels, and outside the 45 min HBOT window identified in Thorn’s rat model of CO reperfusion injury. Clearly, HBOT is effective treatment for CO coma, irrespective of COHb and hypoxia, and it is acting on yet unidentified pathological targets (see Chapter 12). Cerebral arterial gas embolism (CAGE) of diving and non-diving etiology (thousands of cases) similarly was excluded because of the argument that bubbles are the primary pathophysiological target and not ischemia/hypoxia (for discussion see Chapter 11). It has been proposed that most bubbles in CAGE/cerebral decompression sickness have passed the cerebral circulation by the time of HBOT and the primary pathological target of treatment is reperfusion injury which is responsive to HBOT (Harch 1996). In conclusion, the collective experience of HBOT in many of the cases of coma due to CO (especially with delayed treatment 6 h or so) and CAGE is strongly positive and further bolsters the above conclusion on usefulness of HBOT in coma and global ischemia/anoxia.
Another conclusion drawn from this review is that the earlier the HBOT intervention the more impressive the results. In particular, if HBOT is instituted within about 3 h of cerebral insult, over 75% of patients will be noticeably improved or cured. This finding very strongly suggests targets that are both inhibited and stimulated by oxygen. A single hyperacute HBOT greater than or equal to 2 ATA is possibly quenching an on-going injurious cascade, re-energizing stunned neurons similar to the hibernating myocardium reactivation by HBOT (Swift et al1992), and simultaneously reversing any hypoxia or anoxia. The best examples of this are the resuscitation experiments of Van Meter et al (1988, 1999, 2001a, 2001b) and Calvert et al (2002). The Van Meter experiments resuscitated arrested animals 25 min after arrest and truncated brain lipid peroxidation, but had no effect on any white blood cell mediated pathology. The Calvert experiment was a neonatal asphyxia model which inhibited apoptosis. When ischemia is incomplete, prolonged, or treatment is delayed > 45 min HBOT likely inhibits reperfusion injury as demonstrated by the animal data of Thorn, Zamboni, and other models (Harch 2000). As treatment is delayed to 6 h, pressures above 2 ATA are still very effective, but they lose their effectiveness as delays approach 24 h. At this time lower pressures and more treatment are required and suggests treatment of different pathology. Most of the data in this time period derives from TBI studies performed at 1.5 ATA; the results show a dramatic reduction in mortality. With delays longer than one month, HBOT assumes a trophic role stimulating brain repair and possibly manipulating brain blood flow and metabolism as demonstrated in the Harch (1996) animal study, which was replicated in Harch (2001). In both of these experiments (same model, larger numbers in the 2001 experiment) a human low pressure (1.5 ATA) protocol successfully employed from 1990 to 1994 was applied to rats with chronic traumatic brain injury. A series of 80 HBOT treatment improved cognition and increased blood vessel density in the injured hippocampus. This study duplicates the known trophic effect of HBOT in chronic shallow perfusion gradient radionecrosis head wounds (Marx 1990) and likely underpins the mechanism of action of HBOT in the multiple subacute and chronic neurological conditions reported in this chapter.
A more obscure point from this chapter that merits attention is the suggestion of an upper limit to HBOT dosing in chronic brain injury (Harch 2001). Acute oxygen toxicity (overdose) is well known in HBOT such that proper dosing requires a balance of therapeutic benefit with a minimum of negative side effects. Oxygen toxicity in chronic brain injury at pressures< 2 ATA has been considered non-existent in clinical HBOT. The 35 examples in the Harch (2001) article and the case of Miura et al (2002) suggest the opposite in a dose-response fashion. In general this is consistent with the known oxygen toxicity inverse relationship of pressure and duration at pressure, but this finding requires further confirmation by other authors.
HBOT in acute cerebral ischemia/anoxia and coma appears to satisfy the cardinal rule of medicine, prim urn non nocere. In the multitude of cases above and those not reviewed (CO and CAGE) the incidence of serious side-effects of HBOT is surprisingly small. In one review of nearly 1,000 CO poisoned patients (Hampson et al 1994), the maximum seizure frequency was 3% and only occurred at the highest pressures, 2.8-3 ATA, which is greater than the pressure in 49 of the 58 human studies in Table 18.2. The rate dropped ten-fold to 0.3% with pressures of 2.4 ATA. These facts alone argue overwhelmingly for a reasonable attempt, without endangering patients in transport, to perform HBOT in acute cerebral ischemia/anoxia and coma, especially where no other treatment modality exists or has shown clearcut superiority. In essence, HBOT is a simple treatment with potentially profound impact after a single hyperacute administration on devastating incurable neurological conditions that generate monumental long-term tolls of material and human capital and suffering. An increasing number of animal and human experiments/articles are drawing attention to this potential.
HBOT in acute cerebral ischemia/anoxia and coma satisfies the second cardinal rule of medicine- treat until the patient no longer benefits from treatment. In many of the hyperacute and acute studies HBOT benefit was observed on the first or second treatment and was a prelude to further improvement with 1-2 weeks of treatment. The advance in care levels of such patients makes a powerful cost/effectiveness argument from human and financial perspectives. Unfortunately, the tradition in hyperbaric medicine has been to treat once or twice based on the U.S. Navy’s miraculous early results with hyperacute treatment of decompression sickness and air embolism, results not attained equally in the sport scuba diving arena. This stereotyped thinking has subsequently governed the approach to treatment of carbon monoxide poisoning, stroke, and other neurological maladies, ignoring the fact that HBOT initiated at different times after a neurological insult is treating different neuropathology as has been discussed in this chapter. Such an approach may explain the limited positive results of Saltzman ( 11 of 25 patients with temporary improvement after single HBO) and Rockswold ( 47-59% reduction in mortality with 21 treatments, but no long term effect on functional outcome) among others. The greater consideration of the fixed HBOT approach to neurologic injury and the finite, predetermined endpoint of the prospective controlled clinical trial is raised by asking the question of why stop HBOT when the patient is continuing to benefit from treatment or showing neurological gains in the chamber at depth (See Patient 1). New neurological activity at depth strongly suggests ischemic neurological tissue, e.g., ischemic penumbra that would benefit from further HBOT. This arbitrariness is most evident in DCI and CO poisoning where after 5-10 treatments a treating physician is asked how he knows that the patient would not continue to improve on his own. No one knows for sure with each individual case. However, we ask why this factor is not considered at the outset, or after the 2nd, 4th, 7th, 11th, or any other subsequent HBOT treatment when the patient is making stepwise improvement with each or a few HBOT treatments, it is known that it takes one year for a human tissue injury to mature (e.g., wound healing tensile strength and time for neurological injury to be considered chronic), smoldering inflammatory cascades are the underlying pathology, and the analogy of a single HBOT “jump starting a failed engine with a dead battery” is grossly inadequate. In fact, the deciding factor in determining the number of HBOT treatments may be the point in the injury process at which HBOT is initiated, not financial considerations, a factor increasingly dominating medical decision-making in the United States today.
In our accumulating extensive experience, repetitive HBOT appears to be trophic, stimulatory to brain repair, and may not be complete in some cases until 200-300 treatments (see Patient 1). Perhaps the best and most expedient method to assess the HBOT potential and endpoint of treatment at any time after injury for any brain pathology is SPECT brain imaging on a high resolution camera (see Figures 18.1 to 18.6). SPECT before and after any single HBOT at any point in the treatment process may help identify the injured brain’s potential for any or further HBOT. HBOT can then be initiated or continued and combined with multiple other treatment modalities. This approach may help document cost-effectiveness of prolonged HBOT by choosing endpoints. Currently, there are over 600 hyperbaric centers in the United States but less than 12 of these routinely treat the neurological conditions addressed in this chapter with the exception of decompression sickness and CO poisoning. In the last edition of this chapter, we wrote that ”As hyperbaric medicine continues to experience a resurgence in use and expansion of applications and research, the proliferation of chambers and increasing ease of access will facilitate use of HBOT in acute cerebral ischemia/anoxia and coma and will be driven by ever greater lay public and physician knowledge of the above data through widespread computer-assisted dissemination.” These words were prophetic as we are now seeing an Internet driven explosion in HBOT application to chronic neurological conditions. Efforts are currently underway to collect this massive amount of accruing data, publicize the results, leverage scientific proof of efficacy, and achieve reimbursement. The ultimate result will be a retrospective look and application of HBOT to the highest impact targets, acute ischemic/hypoxic brain injury and resuscitation. The outcome will be a large scale dramatic improvement in mortality, morbidity, and quality of life. We predict that this will revolutionize the treatment of brain injury.